- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Cell Signaling Journal
(Discontinued)
ISSN: 1876-3901 ― Volume 4, 2012
Cytokine Deprivation and Cell Death
Paul G. Ekert*, Anissa M. Jabbour
Abstract
It has long been known that many cell types are dependent on specific cytokines that signal proliferation, regulate differentiation and suppress apoptosis. A detailed picture of the structure of several cytokine receptors has added to our understanding of the molecular mechanism of receptor activation. An explosion of knowledge of apoptosis pathways and the ways in which the Bcl-2 family of proteins function has deepened the understanding of the effector arm of the programmed cell death pathway. The challenge is to uncover the molecular links between these two pathways. In this article, we will try to examine what is known of the intersections between cytokine signalling pathways and apoptosis pathways, with particular reference to receptor signalling by the haematopoietic cytokines Interleukin-3 (IL-3) and Granulocyte-Macrophage-Colony Stimulating Factor (GM-CSF).
Article Information
Identifiers and Pagination:
Year: 2011Volume: 3
First Page: 20
Last Page: 26
Publisher Id: TOCELLSJ-3-20
DOI: 10.2174/1876390101103010020
Article History:
Received Date: 2/5/2010Revision Received Date: 21/8/2010
Acceptance Date: 22/10/2010
Electronic publication date: 14/4/2011
Collection year: 2011
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Children’s Cancer Centre, Murdoch Children’s Research Institute, Royal Children’s Hospital and Department of Paediatrics, University of Melbourne, Flemington Rd, Parkville, 3052, Australia; Tel: +613 93455823; Fax: +613 93454993; E-mail: paul.ekert@mcri.edu.au
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 2-5-2010 |
Original Manuscript | Cytokine Deprivation and Cell Death | |
IL-3 AND GM-CSF
IL-3 and GM-CSF are haematopoietic cytokines involved in normal haematopoiesis, including the maintenance and proliferation of myeloid progenitor cells and regulation mye-loid differentiation [1]. GM-CSF and IL-3 have important physiological roles and deregulated signalling contributes to a number of human diseases. In normal physiology, GM-CSF and IL-3 signalling are not required for the develop-ment of a functioning haematopoietic system, but are impor-tant in the expansion or repopulation of certain haematopoie-tic lineages in response to infection or cytopenias, for example after treatment with chemotherapeutic agents [2, 3]. This property underpins the most striking clinical application of GM-CSF (and now more commonly G-CSF) to enhance granulopoiesis after chemotherapy (reviewed in [4]). In Acute Myeloid Leukaemia (AML), overexpression of the IL-3 receptor alpha chain (( chain or CD123) is observed in a subset of leukaemias and is associated with a worse prognosis [5]. Whilst the biological mechanisms by which overexpression of this receptor subunit contributes to the progression of AML remain to be determined, the potential of the ( chain as a therapeutic target, particularly to attack leukaemic stem cells, is being actively explored [6]. The proinflammatory functions of GM-CSF, IL-3 as well as other cytokines may contribute to a number of chronic inflammatory diseases, including arthritis [7]. At least in animal models, depletion or blocking GM-CSF signalling can substantially lessen disease severity, although this has not yet translated into an established therapeutic option in humans.
The receptor for each cytokine is a heterodimeric complex of a common beta chain (ßc) and a cytokine-specific ( chain. The IL-5 receptor also shares the ßc. Loss of IL-3 or GM-CSF signalling in dependent cells results in apoptosis, characterised by mitochondrial outer membrane permeabilisation and displacement of cytochrome c and other factors from mitochondria to the cytoplasm. Cytochrome c acts as a co-factor in the formation of a multimeric complex of the adaptor molecule Apaf-1 and procaspase-9, known as the apoptosome [8]. This complex is the apex of the enzymatic cascade that activates cell death proteases (caspases). Apoptosis is probably a universal response in all cells when they lose essential survival signals. An ability to evade this response by acquisition of mutations or silencing expression of key molecular components contributes to malignant transformation. For example, the translocation associated with follicular lymphoma results in the over-expression of the pro-survival Bcl-2 protein in B-cells that blocks this apoptotic response [9] and in experimental models of malignancy, the inhibition of apoptosis co-operates with other oncogenes to accelerate the development of tumours [10]. In paediatric Acute Lymphoid Leukaemia (ALL), epigenetic mechanisms silence the expression of some proteins required in the normal apoptotic response to cytokine deprivation [11].
BCL-2 FAMILY AND GROWTH FACTOR DEPRIVATION – A HISTORICAL PERSPECTIVE
The WEHI3B cell line is a myeloid leukaemic cell line, in which the primary oncogenic lesions are retroviral insertions leading to the abnormal expression of IL-3 and the homeobox gene HoxB8. Before IL-3 was eventually purified from conditioned media derived from the culture of WEHI3B cells, it was recognised that it could support the growth of bone marrow derived cells. Continuous culture of bone marrow derived haematopoietic cells in WEHI3B conditioned media was used to derive the IL-3 dependent line FDC-P1 [12]. FDC-P1 cells provided a useful tool in the analysis of oncogenes such as Bcr-Abl, which permitted these IL-3-dependent cells to proliferate in the absence of IL-3 [13]. It was in this model that a newly described proto-oncogene, Bcl-2, cloned from the breakpoint of follicular lymphoma, was tested. When Bcl-2 was over-expressed in FDC-P1 cells, these cells were not able to proliferate in the absence of IL-3 as might have been anticipated if Bcl-2 were an oncogene regulating proliferation [14]. Astonishingly however, these cells did not die either. They remained quiescent, arrested in the G0/G1 phase of the cell cycle, but retained the ability to proliferate when IL-3 was restored. The implications were profound; Bcl-2 inhibited the cell death response to IL-3 deprivation and blocking this pathway was oncogenic. Further, IL-3 suppressed the activation of a death pathway (separate from proliferation pathways) that was primed to kill cells should they lose the IL-3 signal.
THE “BCL-2 INHIBITABLE” APOPTOSIS PATHWAY
The Bcl-2 family is subdivided into pro-survival and pro-apoptotic family members. It is the interactions between various Bcl-2 family members that determine whether a cell is configured for survival or death and these are excellently described by Grant Dewson in this edition and have been reviewed elsewhere [15, 16]. Our question for consideration is; how do cytokines, such as IL-3 or GM-CSF, inhibit the activation of the death pathway to keep cells configured for survival, and what happens to cause a switch to death when cytokines are removed? One approach to this question is to identify the Bcl-2 family members involved in regulating apoptosis in response to cytokine degradation, and then determine how cytokine signalling regulates these proteins.
Cells lacking both of the key proapoptoic Bcl-2 family members, Bax and Bak, are completely resistant to IL-3 withdrawal and, like cells over-expressing Bcl-2, proliferate again when growth factor is restored [17, 18]. Regulation of the activation of Bax and Bak is therefore the key step in the commitment to apoptosis. The BH3-only proteins (pro-apoptotic members of the Bcl-2 family containing a single Bcl-2 Homology domain) function as sensors of cellular stresses [19]. They initiate apoptosis by repressing the anti-apoptotic proteins like Bcl-2, Bcl-xL and Mcl-1 and in certain cases directly bind and activate Bax and Bak. One hypothesis to explain how cytokines inhibit cell death is that the activity of particular BH3-only proteins is regulated by cytokine receptor signalling pathways. In such a model, BH3-only proteins are the direct link between cytokine signalling and apoptosis pathways.
WHICH BH3-ONLY PROTEIN?
Experiments in IL-3 dependent cells suggest that three BH3-only proteins, Bad, Bim and Puma, are involved in detecting loss of growth factor signals. Co-immunopreci-pitation experiments demonstrated that Bad was bound to the chaperone protein 14-3-3, in a manner dependent on the phosphorylation of Bad at two critical serine residues by Protein Kinase B (PKB or AKT). Furthermore, dephospho-rylated Bad was no longer able to bind to 14-3-3 and was free to bind to and inhibit the pro-survival protein Bcl-xL [20, 21]. This suggested a model whereby IL-3 signalling, by activating AKT, repressed apoptosis by inactivating Bad. One of the problems with this model is that IL-3 dependent cells derived from Bad-deficient mice remained susceptible to apoptosis provoked by IL-3 deprivation [18]. However, deletion of Bad does contribute to malignant transformation [22], and Bad may act to suppress the development of B-cell lymphomas caused by c-myc over expression [23], but resistance to growth factor deprivation is not likely to be the mechanism.
In contrast, prolonged survival after cytokine deprivation is observed in several cell types derived from both Bim-/- and Puma-/- mice [18, 24-26]. Increased populations of most haematological cell types can be found in the peripheral blood and spleens of Bim deficient mice, presumably as a result of diminished apoptosis. Activated thymocytes from Bim-/- mice survive Interleukin-2 (IL-2) deprivation and Bim-deficient mast cells survive IL-3 deprivation. Puma-deficient myeloid precursor cells or mast cells survive cytokine deprivation and proliferate again when cytokine is restored. These data indicate that both Bim and Puma “sense” the loss of cytokine signalling. The experimental evidence suggests several mechanisms by which this is achieved.
Post-translational modification, specifically phosphory-lation of Bim by kinases activated by cytokine signalling may regulate Bim activity (reviewed in [27]). One Bim isoform, BimEL (Bim “extra long”) is phosphorylated on specific serine residues by ERK1/2 and by JNK. The effect of such post-translational modifications of Bim is to regulate Bim turnover and interactions with other proteins, in general resulting in loss of Bim function. Cytokine signalling directly activates the kinases which, among other substrates, phosphorylate any Bim that may be present and prevent it from promoting apoptosis. This may in part explain how Bim may be expressed at detectable levels in some cells without inducing apoptosis. However, the true physiological role of Bim regulation by phosphorylation in vivo is yet to be established. To date, there is no unequivocal evidence to support or refute post-translational modification of Puma, although potential phosphorylation sites exist within the protein.
Bim and Puma are also transcriptionally upregulated after cytokine deprivation. Perhaps the best established example involves the Forkhead transcription factor FoxO3a [26, 28]. AKT plays central role in this model by phosphorylating FoxO3a and inhibiting translocation to the nucleus in the presence of cytokine signalling. When cytokine signalling is lost, dephosphorylated FoxO3a translocates to the nucleus to induce transcription of Puma and Bim. This model predicts that FoxO3a deficient cells would be resistant to apoptosis provoked by cytokine deprivation. This was directly tested in IL-3-dependent mast cells [26]. Surprisingly, both Puma and Bim protein levels increased after IL-3 deprivation in FoxO3a-deficient cells, but nevertheless some resistance to IL-3 deprivation was observed. However, this resistance to apoptosis was not observed in FoxO3a-/- null myeloid cell lines deprived of IL-3 [29]. In both studies, FoxO3a deletion provided less protection against cytokine deprivation than deletion of Puma, Bim or both.
These data clearly imply other transcription factors also regulate Bim and Puma expression following cytokine deprivation. An increasing body of evidence suggests that p53 transcriptional activity is activated in the response to cytokine deprivation, at least in some cell types. This was recognised in untransformed myeloid precursor cells derived from p53-/- mice which had increased survival in limiting cytokine concentrations compared to wild type cells [30]. This was confirmed in primary myeloid progenitor cells and transformed, IL-3 dependent cell lines [29, 31]. P53-dependent regulation of Puma transcription is critical to the mechanism by which p53 induces apoptosis after cytokine withdrawal, however, it is less clear how p53 transcriptional activity itself is activated by cytokine deprivation. One possible link between the cytokine receptor signals and p53 again implicates AKT [29, 32, 33]. AKT is sufficient to phosphorylate and activate MDM2, the E3 ubiquitin ligase responsible for p53 degradation. In the presence of AKT activation, p53 transcriptional activity is suppressed because less p53 is available to translocate to the nucleus. After cytokine deprivation, MDM2 is itself degraded, in effect increasing the pool of free active p53 able to initiate transcriptional activity. Studies in AKT-deficient cells will establish the veracity and physiological significance of this pathway.
OTHER BCL-2 FAMILY MEMBERS
Deletion of Puma or Bim or both does not provide the same survival and retention of clonogenic potential as deletion of both Bax and Bak [18, 26]. In contrast, over-expression of anti-apoptotic Bcl-2 family members such as Bcl-2, Bcl-xL or Mcl-1 does “phenocopy” Bax/Bak double knockout cells. Thus although cytokine signalling represses the activation of certain BH3-only proteins, maintenance of anti-apoptotic Bcl-2 protein levels is also an efficient way to neutralise all active BH3-only proteins and critical to prevent the activation of Bax and Bak after cytokine deprivation. The best example of regulation of anti-apoptotic Bcl-2-like proteins by cytokine receptor signalling is Mcl-1. Mcl-1 protein levels are tightly regulated by GM-CSF and IL-3 signalling. Loss of GM-CSF receptor signalling results in rapid, proteasomal degradation of Mcl-1 [34], which implies that GM-CSF receptor signalling is maintaining Mcl-1 at levels sufficient to prevent the activation of Bax and Bak. In this instance, the kinase responsible for Mcl-1 phosphorylation (which targets Mcl-1 for proteasomal degradation) is Glycogen Synthase Kinase-3 (GSK-3). GSK-3 itself is negatively regulated by phosphorylation by AKT activated by IL-3 signalling [35]. Thus, in the presence of IL-3 or GM-CSF signalling, AKT activation suppresses GSK3 which maintains Mcl-1 levels. More recent data indicate that transcriptional regulation of Mcl-1 levels by mTOR-dependent mechanisms are important in the development tumours that arise as a result of constitutive activation of AKT signalling [36]. The contribution this mechanism makes to normal survival signals transduced by cytokines such as IL-3 and GM-CSF remains to be determined, but is clear that AKT and mTOR activation are normal consequences of IL-3 and GM-CSF receptor signalling.
RECEPTOR ACTIVATION
IL-3 and GM-CSF receptors do not have intrinsic tyrosine kinase activity. Instead, a Janus kinase, JAK-2, is recruited to the ßc and is phosphorylated following ligand-receptor binding. In turn, JAK-2 phosphorylates tyrosine residues on ßc and initiates several signalling cascades. The solution of the structure of the GM-CSF receptor has provided insights into the activation of the receptor by ligand binding [37]. The GM-CSF receptor consists of a hexameric complex of the four domains of a common ß chain and the two domains of a GM-CSF receptor ( chain. However, a higher order, dodecameric complex is the active signalling conformation. Signalling from this conformation is critically dependent on the formation of an interaction surface by two ßc and one the ( chains (termed site 4). In the dodecameric conformation, the JAK-2 molecules are sufficiently close to allow transphosphorylation to occur. Mutations to this surface disrupt dodecamer formation but not hexamer formation, and significantly block proliferative signalling.
SIGNALLING KINASES
The phosphorylated tyrosines in ßc act as docking sites for adaptor molecules involved in the initiation of other signalling pathways, including the activation of PI3K/AKT, RAS/RAF/ERK or JAK/STAT pathways. Mutational analyses of the ßc have mapped regions and specific tyrosine residues in ßc required for the activation of specific signalling pathways and cell-biological responses [38-40] (Fig. 1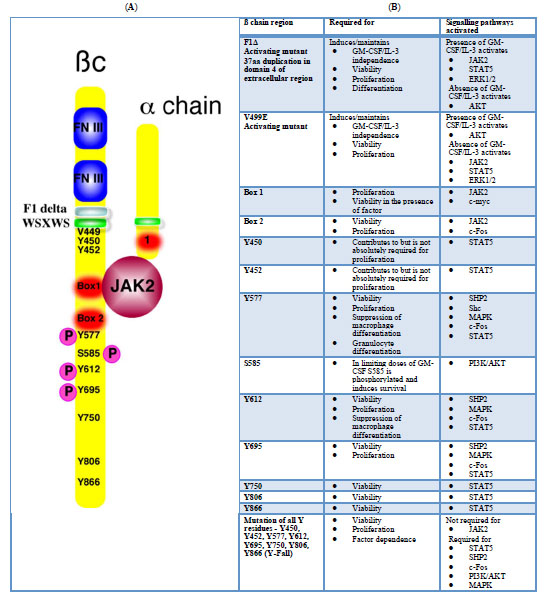 ). Mutations which abolish JAK-2 binding or activation prevent proliferative and survival signalling. It is worth noting, however, that deletion of JAK-2 abolished proliferative responses to IL-3 signalling in haematopoietic stem cells, but these stem cells were still present and the proliferative response could be rescued by retroviral infection of JAK-2 [41]. This indicates that JAK-2 is not required for the development or survival of at least some haematopoietic progenitor cells which could be made IL-3 responsive. Y577, Y612 and Y695 in JAK-2 were sufficient to signal proliferation. Observations in human GM-CSF responsive cell lines identified that phosphorylation of a conserved serine residue, S585, was critical for suppressing apoptosis, particularly at limiting concentrations of cytokine [42]. At low doses cells survived but did not proliferate and ßc was phosphorylated at S585 but not at Y577. With increased GM-CSF doses, Y577 phosphorylation was detected, cells began dividing but S585 phosphorylation was lost. Limiting GM-CSF concentrations may be sufficient to maintain survival of haematopoietic stem cells but proliferation occurs only when abundant cytokine is present, perhaps at times of infection or in response to neutropenia. If the concentration of GM-CSF can determine whether a survival or proliferative signal is transduced, several interesting questions present themselves as to how to account for this. Is the receptor in a different confirmation at low dose GM-CSF compared to high dose GM-CSF? If the conformation is dodecameric, why is there no tyrosine phosphorylation? Can the receptor in the hexameric conformation transduce a survival signal? If this were the case, this signal would presumably be independent of tyrosine kinase activity.
). Mutations which abolish JAK-2 binding or activation prevent proliferative and survival signalling. It is worth noting, however, that deletion of JAK-2 abolished proliferative responses to IL-3 signalling in haematopoietic stem cells, but these stem cells were still present and the proliferative response could be rescued by retroviral infection of JAK-2 [41]. This indicates that JAK-2 is not required for the development or survival of at least some haematopoietic progenitor cells which could be made IL-3 responsive. Y577, Y612 and Y695 in JAK-2 were sufficient to signal proliferation. Observations in human GM-CSF responsive cell lines identified that phosphorylation of a conserved serine residue, S585, was critical for suppressing apoptosis, particularly at limiting concentrations of cytokine [42]. At low doses cells survived but did not proliferate and ßc was phosphorylated at S585 but not at Y577. With increased GM-CSF doses, Y577 phosphorylation was detected, cells began dividing but S585 phosphorylation was lost. Limiting GM-CSF concentrations may be sufficient to maintain survival of haematopoietic stem cells but proliferation occurs only when abundant cytokine is present, perhaps at times of infection or in response to neutropenia. If the concentration of GM-CSF can determine whether a survival or proliferative signal is transduced, several interesting questions present themselves as to how to account for this. Is the receptor in a different confirmation at low dose GM-CSF compared to high dose GM-CSF? If the conformation is dodecameric, why is there no tyrosine phosphorylation? Can the receptor in the hexameric conformation transduce a survival signal? If this were the case, this signal would presumably be independent of tyrosine kinase activity.
 |
Fig. (1) A. The GM-CSF/IL-3/IL-5 receptor is a heterodimer of a specific alpha chain and a common beta chain (ßc). The extracellular domain of the ßc has two fibronectin type III repeats involved in IL-3/GM-CSF binding. The ßc also has a WSXWS transmembrane domain. Activating mutations of ßc have been identified in the transmembrane domain (e.g. V449E and F1Δ [40]). The cytosolic portion of ßc contains a Box 1 and Box 2 motifs, involved in JAK binding and activation. Deletion of these domains abolishes signalling. A number of tyrosine residues are phosphorylated after ligand binding. Tyrosines (Y) 577, 612 and 690 are all involved in the transduction of proliferative signals in addition to some survival signals). Mutations Y577F or Y612F abolish signalling and each residue alone is sufficient for STAT5 and Shp phosphorylation. The other tyrosine residues, when individually mutated to phenylalanines, also result in diminished proliferation. Serine 585 is phosphorylated at limiting doses of cytokine and transduces a survival signal. B. The table highlights the various signal transduction cascades that are activated by various regions of the ß#x00DF;c. The references from which this table was compiled are [38-40, 47-49] |
There are several cytosolic signalling pathways activated when IL-3 or GM-CSF binds to their respective receptors which have been described to regulate cell survival via a variety of mechanisms (Fig. 2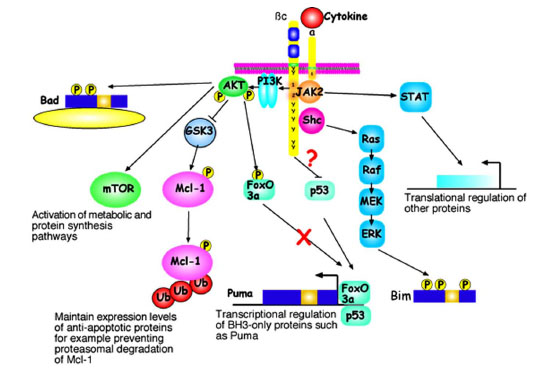 ). Although represented separately, there is considerable cross-talk between these pathways which makes assignation of specific biological responses to particular signalling cascades challenging. For example, Ras activation can activate PI3K signalling and PI3K/AKT and Ras pathways activate downstream targets such as mTOR. Much focus has centred on the activation of the PI3K/AKT pathway as the principal “survival pathway”. Potential substrates have already been mentioned, including BH3-only proteins and FoxO3a. Other potential pro-survival functions of AKT may be mediated viaeffects on maintaining nutrient uptake and protein synthesis through the activation of proteins including mTOR and S6 kinase. Further, AKT may regulate a p53-dependent response to IL-3 deprivation [29]. However, it may be a significant overstatement to describe the PI3K/AKT pathway as the “survival pathway”. Enforced expression of constitutively active AKT does prolong the survival of myeloid cells in the absence of IL-3, however, at least half such cells die in the first days of the experiment [43]. In our hands, AKT over-expression delays but does not prevent apoptosis after IL-3 withdrawal (unpublished data).
). Although represented separately, there is considerable cross-talk between these pathways which makes assignation of specific biological responses to particular signalling cascades challenging. For example, Ras activation can activate PI3K signalling and PI3K/AKT and Ras pathways activate downstream targets such as mTOR. Much focus has centred on the activation of the PI3K/AKT pathway as the principal “survival pathway”. Potential substrates have already been mentioned, including BH3-only proteins and FoxO3a. Other potential pro-survival functions of AKT may be mediated viaeffects on maintaining nutrient uptake and protein synthesis through the activation of proteins including mTOR and S6 kinase. Further, AKT may regulate a p53-dependent response to IL-3 deprivation [29]. However, it may be a significant overstatement to describe the PI3K/AKT pathway as the “survival pathway”. Enforced expression of constitutively active AKT does prolong the survival of myeloid cells in the absence of IL-3, however, at least half such cells die in the first days of the experiment [43]. In our hands, AKT over-expression delays but does not prevent apoptosis after IL-3 withdrawal (unpublished data).

Other kinases also promote prolonged survival after cytokine deprivation. Pim-2 is a serine/threonine kinase transcriptionally down-regulated following IL-3 withdrawal. Much like AKT, enforced expression of Pim-2 results in prolonged survival of at least some cells after IL-3 deprivation [43]. The related kinase Pim-1 may also transduce survival signal in basophils that permits survival in the absence of IL-3 [44]. Over-expression of an activated form of Ras blocked IL-3 withdrawal induced apoptosis without inducing significant IL-3 independent proliferation in an IL-3 dependent cells line [45]. Activating Ras mutations that occur in Juvenile Myelomonocytic Leukaemia (JMML) are responsible for the characteristic ability of leukaemic cells with these mutations to proliferate (and survive, of course) in growth factor-free (GM-CSF) culture conditions. MAP Kinase activity also contributes to IL-3 dependent proliferation and survival signalling [46]. There are clearly several important signalling pathways that contribute to the survival and proliferative effects of IL-3 and GM-CSF receptor signalling. How easily and specifically these pathways may be subdivided into survival or proliferation pathways is of considerable interest. It is self-evident that survival signalling is essential for all the normal function of cytokines. Survival signals also contribute to the pathogenesis of diseases such as acute myeloid leukaemia [42]. It would be naive to think that regulation of survival by cytokines will depend on a single signalling pathway or a sole Bcl-2 family member. The complexity of these interacting pathways means there is still much to be learned.