- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Cell Signaling Journal
(Discontinued)
ISSN: 1876-3901 ― Volume 4, 2012
Dying for Something to Eat: How Cells Respond to Starvation
Alfredo Caro-Maldonado, Cristina Muñoz-Pinedo*
Abstract
Food is a requirement of life. Unicellular and multicellular organisms have therefore developed mechanisms to detect, react to and, if necessary, survive the lack of it. In mammals, responses to lack of nutrients in blood are coordinated at the organismal level by hormonal cues. However, individual cells also sense and respond to nutrient deprivation, which occurs under physiological or pathological situations such as fasting, ischemia or solid tumor development. Frequently cells try and survive nutrient deprivation by reducing their energy and carbon requirements and by recycling structural components. However, under certain conditions, the cell reacts to nutrient deprivation by engaging the mitochondrial pathway of apoptosis. Indeed, the metabolic state of the cell can regulate pro- and antiapoptotic Bcl-2 family proteins such as PUMA, Bad, Bim or Mcl-1, thus altering the response to pro-apoptotic stimuli. Severe energetic stress can also kill cells by a form of death with necrotic phenotype. Metabolic pathways are currently receiving enormous attention from cell biologists, due to the fact that tumors have a special metabolism which makes them more susceptible to lack of specific nutrients, particularly glucose and glutamine. For this reason, targeting tumor metabolism opens new therapeutic avenues. We will discuss how nutrient deprivation engages cell death pathways, and how cell metabolism interferes with the apoptotic machinery by regulating apoptotic proteins. We will also re-examine the hypothesis that ATP levels determine whether a cell dies by apoptosis or necrosis. Finally, we will discuss how and why metabolic stress can lead to either cell adaptation and survival or cell death
Article Information
Identifiers and Pagination:
Year: 2011Volume: 3
First Page: 42
Last Page: 51
Publisher Id: TOCELLSJ-3-42
DOI: 10.2174/1876390101103010042
Article History:
Received Date: 2/5/2010Revision Received Date: 21/8/2010
Acceptance Date: 22/10/2010
Electronic publication date: 14/04/2011
Collection year: 2011
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http: //creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the IDIBELL, Hospital Duran i Reynals 3ª Planta, Gran Vía de L’Hospitalet 199, L’Hospitalet, Barcelona, 08907, Spain; Tel: 93 260 7339; Fax: 93 260 7426; E-mail: cmunoz@idibell.cat
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 2-5-2010 |
Original Manuscript | Dying for Something to Eat: How Cells Respond to Starvation | |
NUTRIENT DEPRIVATION IN PHYSIOLOGICAL AND PATHOLOGICAL SITUATIONS
Living organisms are continuously subjected to periods of starvation in nature. Nutrient availability determines the expansion of species and colonization of new ecological niches. Nutrients also dictate the appropriate time for mating, sporulating, hibernation and differentiation, from unicellular photosynthetic organisms to mammals. Humans suffer transient or persistent starvation due to a lack of food intake, either because of voluntary dieting, night fasting, or due to the scarcity of available food. Starvation is linked to our first contact with the world, since there is a period of mild starvation in the newborn before breastfeeding is established.
At the cellular level it is possible to observe pathological starvation during ischemia, which is a severe form of starvation due to simultaneous nutrient and oxygen deprivation. Cells in solid tumors are also very commonly exposed to starvation, because the tumor mass grows faster than the blood vessels. This leads to the formation of “necrotic cores”: areas that contain dead cells which did not get enough blood irrigation, and have not been able to survive oxygen and nutrient deprivation [1].
During times of scarcity, unicellular organisms try to save energy by undergoing specialized forms of hibernation such as sporulation. Multicellular organisms usually have specialized cells that detect nutrient deprivation –such as pancreatic cells in animals-, and specialized storage tissues such as adipose tissue. In response to starvation, animals use hormonal cues to mobilize stored resources to provide nutrients to individual cells. Besides whole-body responses to nutrient deprivation -such as the sensation of hunger which drives us to search for food-, individual cells sense and react to lack of nutrients. At the cellular level, starvation of specific nutrients triggers different responses in different tissues, since some tissues like the brain prefer glucose as a source of energy, while other tissues such as the heart or resting muscles prefer fatty acids as fuel. Nutrient deprivation at the cellular level usually leads to mobilization of stored nutrients and concomitant reduction of cellular functions. However, under some circumstances, cells are not able to adapt to stress, and metabolic or energetic stress leads to apoptosis or necrosis.
Apoptosis is a form of cell death which is activated in animals in response to cell-damaging agents, or to developmental cues1. The fact that many human cells die by apoptosis when deprived of a particular nutrient is intriguing. Cells die by apoptosis when they are not needed anymore (for instance after an immune response, when the pathogen has been eliminated), or when they are injured (for instance after a DNA-damaging insult or viral infection2). When cells are deprived of nutrients, their primary response is usually to stop cycling (if they were proliferating), and try to survive using minimal amounts of energy and building blocks. It is thus still unknown why and how some cells engage the apoptotic machinery instead of arresting and waiting for “better times”. Moreover, cells which have undergone tumorigenic transformation are more prone to die than their normal counterparts. We will discuss how the decision of undergoing apoptosis instead of stopping the cycle might be influenced by oncogenes, and can thus be exploited for cancer treatment.
NUTRIENT DEPRIVATION IS SENSED BY AMPK AND mTOR AND LEADS TO CELL CYCLE ARREST, INHIBITION OF PROTEIN SYNTHESIS AND AUTOPHAGY
Cells require four types of “building blocks” to grow: lipids (fatty acids) to make membranes, amino acids for protein synthesis, nucleotides to build nucleic acids, and sugars to produce ATP and multiple macromolecules. Blood provides all of these nutrients to cells in our bodies, and researchers provide these nutrients to cells in culture in the form of enriched culture medium plus serum from animal blood. Since cells need to maintain the equilibrium between catabolism and anabolism, they need to sense the energetic state of the cell and react to starvation. We could define starvation at the cellular level as the lack of any nutrient that the cell requires in order to perform their functions. Unfortunately, the term ‘starvation’ is used in the scientific literature to describe many different stimuli which may lead to different outcomes. We can “starve” cells by incubating them in the simultaneous absence of glucose, amino acids, oxygen and serum: a condition that resembles ischemia. But “starvation” is also used to describe deprivation of only one type of nutrient. For example, in autophagy studies “starvation” is very frequently used to describe amino acid deprivation. Thus, the term starvation is too broad and imprecise when we describe cell responses to the lack of a specific nutrient, since the responses to the absence of each of these nutrients might be completely different. A cell senses the presence of glucose, amino acids, fatty acids or oxygen in different manners, and it reacts differently to the lack of each of them. Moreover, “starvation” or “nutrient deprivation” can lead to energetic stress.
The term “energetic” or “energy stress” has also been used frequently to define any type of stress that would lead to a loss of ATP, which is our measurement of energy levels. As we will discuss later, it is the current view that loss of ATP would convert an apoptotic phenotype to a necrotic one [2]. It should be understood however that nutrient deprivation does not necessarily lead to energetic stress, and energetic stress is not necessarily caused by nutrient deprivation. Thus, deprivation of oxygen or one specific nutrient does not necessarily lead to an energetic stress, while it still triggers a hypoxic response or a starvation response specific for that particular nutrient. Responses are also particularly complex due to the fact that amino acids, glucose and fatty acids serve both as building blocks and cell-type specific energy donors.
Cells detect levels of energy or nutrients by sensing ATP and amino acid levels. The signaling pathways involved in nutrient/energy sensing primarily lead to cell adaptation. When normal nutrient flux is insufficient for the metabolic needs of the cell, the cell must decrease its needs. Additionally, the cell must degrade storage molecules such as glycogen, and if necessary, undergo autophagy. Autophagy is a homeostatic process that consists in the engulfment of cytosolic proteins and organelles into vesicles that fuse to lysosomes. This allows cells to degrade cellular components that serve as structure but also as an energy reserve.
Lack of glucose or nutrients which a cell needs in order to maintain ATP, leads to the activation of AMPK (SNF1/AMP-activated protein kinase) [3]. AMPK is a trimeric protein complex with kinase activity which is allosterically activated by AMP. Mammalian AMPK is sensitive to the cellular AMP:ATP ratio, which is a good indicator of the energetic state of the cell. Activation of AMPK allows the cell to react to low ATP by regulating processes like glucose transport. This is achieved by increasing translocation of a glucose transporter to the plasma membrane. Moreover, AMPK regulates transcription, inhibiting promoters of glucose-repressed genes and downregulating expression of biosynthetic genes. Low ATP levels and AMPK activation also inhibit cell proliferation through the activation of p53 and stabilization of the cell cycle inhibitor p27 [4, 5].
Another key element for the maintenance of energy homeostasis is TORC1. In mammals, TORC1 is a multiprotein complex that contains the mammalian target of rapamycin (mTOR). mTOR promotes protein synthesis, and it is highly active in proliferating cells. It is activated by hormones and growth factors, and it is also stimulated by intracellular amino acids. As recently reviewed by Wang et al. [6], it is not yet known whether amino acids activate mTOR directly or indirectly. A few proteins which play a role in allowing mTor to sense amino acid levels are MAP4K3 [7] and Rag A and B [8]. But their exact role in the pathway requires further studies. Ultimately, lack of glutamine and leucin results in inhibition of mTor, which in turn leads to inhibition of protein synthesis.
mTORC1 regulates protein synthesis by directly phosphorylating two types of proteins that modulate components of the translational machinery: S6 kinases (S6Ks) and the eukaryotic initiation factor (eIF) 4E-binding protein (4E-BPs) [6]. Additionally, amino acid starvation and mTOR inhibition decrease cdc2 activity thus stopping cell cycle. mTOR inhibition also induces autophagy, which, as described above, helps cells survive under low nutrient conditions.
AMPK activation can also lead to the indirect inactivation of mTOR. Thus, a reduction in either ATP levels or amino acid levels will ultimately lead to inhibition of protein synthesis, cell growth arrest and induction of autophagy (Fig. 1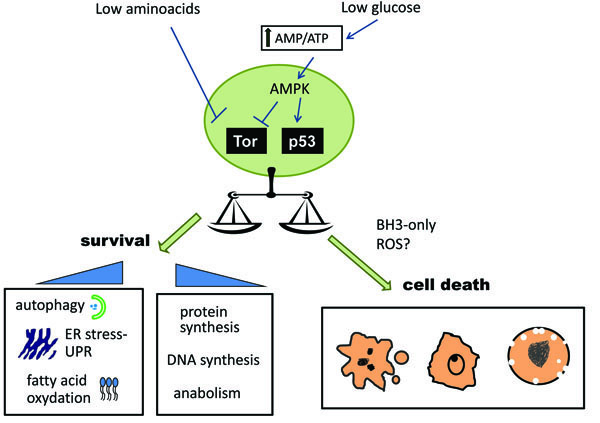 ). In summary, one cellular response when cells are confronted with lack of nutrients is to shift to catabolism, thereby staving off an energetic collapse that could lead to cell death.
). In summary, one cellular response when cells are confronted with lack of nutrients is to shift to catabolism, thereby staving off an energetic collapse that could lead to cell death.

LACK OF NUTRIENTS REGULATES BCL-2 PRO-TEINS
Caspases are proteases that are the effector enzymes for the apoptotic program. Apoptotic cells are readily phagocy-tosed and apoptosis therefore limits extracellular release of cytoplasmic content, which can induce inflammation [9]. There are two main pathways to apoptosis. The extrinsic pathway3, is induced by extracellular proteins termed ‘death ligands’. The intrinsic or mitochondrial pathway is mediated by the apoptosome, a protein complex formed by caspase-9, Apaf-1 and the mitochondrial protein cytochrome c. This complex is formed upon the release of cytochrome c from the mitochondrial intermembrane space, an event that requires the proteins Bax and Bak, which oligomerize and form a pore in this membrane. This process is regulated by other Bcl-2 family proteins like the anti-apoptotic Bcl-2/Bcl-xL or Mcl-1, and the proapoptotic “BH3 only” such as Bim, Bid, Bad or PUMA [10]4.
A number of studies have indicated that death induced by nutrient deprivation, and especially by inhibition of glucose metabolism, is mediated by the mitochondrial apoptotic pathway in many different cell types (Table 1). For instance, low glucose levels induce Bax translocation, a drop in mitochondrial membrane potential and cytochrome c release in hematopoietic FL5.12 cells [11]. Moreover, glucose withdrawal-induced death can be inhibited by Bcl-2 in MCF-7 cells [12] or by Bcl-xL in Baf3 [13].

Recently, a number of studies have shown the importance of the amino acid glutamine as a tumor-specific nutrient. Transformation by the oncogene myc drives tumor progression, but it also confers sensitivity to glutamine depletion [14]. So far, only a few studies have provided insight into death induced by glutamine deprivation [15, 16]. Myc-transformed fibroblasts have been shown to die through the intrinsic pathway of apoptosis when deprived of glutamine because overexpression of Bcl-2 or a dominant-negative form of caspase-9 prevented death [15]. The authors also demonstrated that caspase-8, a caspase implicated in the extrinsic pathway, was not involved, since the expression of a viral inhibitor of caspase-8 did not prevent death.
Mitochondrial apoptosis is regulated by BH3-only proteins, which contain a Bcl-2 homology motif (the BH3 motif) which promotes activation of Bax and Bak leading to cytochrome c release. Amongst these proteins, Bad has been extensively linked to glucose metabolism and glucose deprivation-induced cell death [17]. Bad has been recently shown to behave as a “moonlighting” protein, serving a role in cell death but also in glucose metabolism. Bad is present in a complex in the mitochondria which includes the glycolytic enzyme glucokinase (hexokinase IV). This complex displays glucose phosphorylating activity and helps provide mitochondria with metabolic intermediates for ATP production [18]. Moreover, Bad participates in glucose sensing and blood glucose clearing. Bad-deficient mice develop glucose intolerance, and isolated pancreatic islets from Bad deficient animals secreted significantly lower amounts of insulin when perfused with glucose [19].
Involvement of Bad in glucose deprivation-induced cell death is supported by a few experiments. Treatment of human leukemic cells with a glycolytic inhibitor led to Bad dephosphorylation and BAX oligomerization on mitochon-dria [20]. Moreover, analysis of Bad-deficient mice revealed that Bad mediates apoptosis induced by glucose deprivation in mouse hepatocytes [18]. These experiments suggest that Bad serves as a BH3-only protein to directly mediate mitochondrial outer membrane permeabilization. However, the fact that this protein is involved in glucose sensing and metabolism complicates the interpretation of these results. The same region of the protein, the BH3 domain, mediates both functions of the protein: glucose metabolism and cell death. It remains to be determined whether the implication of Bad in glucose withdrawal-induced apoptosis is direct, through its effect on Bcl-2 family proteins, or indirect, through its effects on glycolysis, or both.
PUMA is a BH3-only protein which is strongly induced in response to DNA damage and growth factor withdrawal5. It has been recently described that PUMA is induced by glucose deprivation in murine hematopoietic cells [21]. Downregulation of PUMA with siRNA was sufficient to reduce death induced by glucose deprivation, indicating that PUMA mediates glucose withdrawal-induced cell death. PUMA can be induced in a p53-dependent manner after DNA damage and growth factor withdrawal [22]. Zhao et al. showed that PUMA induction after glucose deprivation was also mediated by the transcription factor p53 [21]. Moreover, downregulation of p53 conferred a moderate protection from cell death, consistent with the moderate downregulation of PUMA and p53 levels that could be achieved by RNAi. Intriguingly, however, although p53 was activated, its levels were not altered upon glucose deprivation. Therefore, cell death after glucose deprivation seemed to be dependent on PUMA induction and p53 activity, but not on induction of p53.
Bim is one of the best studied BH3 proteins. Bim is essential for the execution of some of the apoptotic stimuli that can be antagonized by Bcl-2, including growth factor withdrawal and deletion of autoreactive T cells [23]. It was recently determined that Bim is responsible for activation of the mitochondrial pathway in some cell lines after endoplasmic reticulum (ER) stress [24]. This suggests that Bim may be involved in death induced by nutrient deprivation, since one of the consequences of nutrient deprivation is ER stress. Glucose is necessary for protein glycosylation, and amino-acids are necessary for protein synthesis. Malfunctioning of protein glycosylation or synthesis can cause accumulation of misfolded proteins, which leads to ER stress. For this reason, it is possible that ER stress, rather than ATP deprivation, is the cause for many of the consequences of glucose deprivation, including cell death. Supporting this idea, it has been demonstrated that the glucose analog 2-deoxyglucose kills some cell lines due to its effects on protein glycosylation rather than on ATP synthesis [25].
Bim is induced by ER stressors, and it was recently shown that Bim is also induced upon glucose withdrawal in murine hematopoietic cells, possibly due to ER stress caused by impairment of glycosylation [21]. Moreover, Bim mediates glucose deprivation-induced cell death of these cells, as revealed by the use of RNAi. It was not reported whether induction of Bim in this system was due to transcriptional or posttranscriptional regulation; it is possible that both levels of regulation were altered, since ER stress can induce both stabilization of the protein and transcription of Bim mRNA [24]. Interestingly, the same transcription factor involved in upregulation of Bim after ER stress, CHOP, is induced by amino acid deprivation [26]. This suggests that upregulation of Bim could be the cause for apoptosis induced by lack of several types of nutrients: glucose or amino acids.
Noxa is another BH3-only protein which can mediate apoptosis induced by either ER stress [27] or by glucose deprivation. Noxa was shown to be involved in apoptosis of Jurkat cells after glucose withdrawal, and downregulation of Noxa conferred a competitive survival advantage to primary T cells under low glucose conditions [28]. The authors hypothesized that the role of Noxa in this setting was related to its ability to neutralize the antiapoptotic Bcl-2 homolog Mcl-1, and when they generated Jurkat cells with low levels of Mcl-1, they observed that these cells were more sensitive to glucose deprivation than the wild type cells. Thus, Mcl-1, like Bcl-2 and Bcl-xL, can protect cells from glucose deprivation-induced apoptosis. Moreover, as we will discuss later, Mcl-1 is regulated by glycolytic metabolism, which suggests that downregulation of Mcl-1 may be relevant for apoptosis induced by absence of glucose in different systems [29].
NUTRIENT DEPRIVATION ALSO TRIGGERS NEC-ROSIS AND CASPASE-8 DEPENDENT APOPTOSIS
Apoptosis has been defined as “controlled cell death”, in contrast to necrosis that was described as a form of uncontrolled death that leads to the release of cytoplasmic content. Recently it has been described that necrosis is not just a passive process, but there are molecular mechanisms that are regulated during some forms of necrosis. For instance, necroptosis (RIP dependent necrosis), is a form of caspase independent, RIP kinase-dependent cell death [30]. Golstein and Kroemer reviewed the events that frequently occur during necrosis, some of which are related with mitochondrial dysfunction and can also occur during apoptosis: ATP depletion, mitochondrial swelling, cytoplasmic Ca2+ increase, Reactive Oxygen Species (ROS) production, activation of proteases like calpains, and finally, membrane rupture [31].
As discussed above, cells frequently die by apoptosis through the mitochondrial pathway when deprived of nutrients. However, many tumors have acquired resistance to the mitochondrial pathway through the overexpression of the pro-survival Bcl-2, Bcl-xL or Mcl-1, or mutations in pro-apoptotic BH3-only proteins or Bax and/or Bak. Bax and Bak are Bcl-2 family proteins required for cytochrome c release, and cells which are deficient in Bax and Bak cannot undergo apoptosis upon treatment with inducers which kill through the mitochondrial pathway (See review by Grant Dewson in this issue). In the absence of Bax and Bak, some apoptotic stimuli trigger necrosis. E. White and colleagues studied death induced by metabolic stress (glucose deprivation under hypoxic conditions) in Akt-overexpressing mouse baby kidney cells [32]. They showed that when subjected to metabolic stress, Bax, Bak-deficient cells died by necrosis. Conversely, their wild-type counterparts died by apoptosis, indicating that metabolic stress induces apoptosis when Bax and Bak are present, but necrosis in cells which cannot die by the mitochondrial pathway. Intriguingly, however, a recent report indicated that mouse embryonic fibroblasts (MEFs) deficient in Bax and Bak die by apoptosis in response to glucose deprivation under normoxia [33]. These cells bypassed the mitochondrial pathway by engaging caspase-8, which is the apical caspase in the death receptor pathway of apoptosis. Unfortunately, the caspase-8 activating complex was not identified. Caspase-8-dependent apoptosis in response to glucose deprivation is not restricted to MEF: this protease is also involved in death of HeLa cells upon glucose starvation. Since HeLa cells die through the mitochondrial pathway in response to many stimuli, these results suggest that caspase-8 may mediate death induced by glucose deprivation in other cell types, perhaps through direct cleavage of caspase-3, or through the cleavage of the BH3-only protein Bid which could trigger the release of mitochondrial pro-apoptotic proteins.
Human adult cardiomyocytes downregulate many proteins of the apoptotic machinery during development, including Apaf-1, Bax and caspases. Cardiomyocytes were shown to die in a necrotic manner during experimental ischemia, which was achieved by incubating cells in a saline solution under hypoxia [34]. Since heart and brain ischemia (stroke) kill millions of people every year, it will be of great relevance to understand the mechanism of this form of caspase-independent cell death. ROS are common mediators of ischemic cell death, suggesting that oxidation of cellular components is the cause for necrosis, in the presence or absence of the apoptotic machinery [35]. It is well known that nutrient deprivation frequently triggers generation of Reactive Oxygen Species (ROS), since the lack of certain metabolic intermediates impairs the production of antioxidants like gluthatione or NADPH [36]. Thus, under some circumstances, cells may die of necrosis upon ischemia or nutrient deprivation due to massive ROS production. It remains to be determined what tips the balance towards apoptosis or necrosis in cells with an intact apoptotic machinery.
Another example of non-apoptotic cell death induced by metabolic stress was described by Yuneva et al. [15], who observed that glucose withdrawal induced cell death in untransformed human fibroblasts. This form of death did not display classical chromatin condensation, and was not inhibited by the antiapoptotic protein Bcl-2. While caspase activation was not looked at in this model, the authors suggested that this form of death was not apoptotic. However, it is possible that in this system, as in Bax, Bak deficient MEFs, caspase-8 mediates cell death. Cell death without classical chromatin condensation was observed after subjecting human HepG2 hepatoma cells to glucose deprivation [37]. These authors also used the term ‘necrosis’ to describe this form of cell death, as neither Annexin binding or nuclear condensation could be observed. However, caspase inhibitors prevented cell death, and death was accompanied with caspase activation which shows that, by definition, this death was apoptotic. Thus in the absence of glucose, cells can die in a manner which is dependent on caspases but which can be mistaken with necrosis due to the lack of classical chromatin condensation. Indeed, Caro-Maldonado et al. reported atypical, non-apoptotic chromatin condensation upon glucose deprivation in Bax, Bak-deficient MEFs, although these cells showed all other hallmarks of apoptosis [33].
As a summary, we can conclude that nutrient deprivation can induce apoptosis, necrosis, and caspase-dependent cell death without some classic apoptotic features, such as chromatin condensation (Fig. 1 and Table 1). The factors that determine whether cells die by apoptosis or necrosis are still unknown. It has been suggested that ATP depletion can promote a switch to necrotic cell death in response to certain pro-apoptotic stimuli. Apoptosis is considered to be an ATP-dependent process and frequently described as “energy dependent death” while necrosis is thought to be a more ‘passive’ form of death. For this reason, it could be hypothesized that nutrient deprivation could induce apoptosis or necrosis depending on how much ATP is depleted by nutrient removal. However, the assumption that apoptosis requires ATP remains to be formally proven, since so far it is not possible to deplete ATP from a whole cell without altering many other parameters. Original experiments that led to the hypothesis of requirement of ATP for apoptosis were performed in Jurkat cells which were incubated with an inhibitor of respiration in the absence of glucose. Under these ATP depleting conditions, apoptosis inducers such as staurosporine or a death ligand triggered a necrotic death without apoptotic features [38-40]. Thus, it was concluded that ATP depletion leads to a switch from apoptosis to necrosis. However, ATP depletion is only one of the many consequences of the simultaneous inhibition of respiration and glycolysis. This severe form of starvation could potentially lead to a reactive oxygen species burst which could be responsible for the switch between apoptosis and necrosis. Moreover, since tumor cells frequently make fatty acids and nucleotides from glucose, it remains possible that apoptosis was inhibited due to the loss of certain lipids or nucleotides, and not as a consequence of a drop in ATP.
In vitro studies had shown that formation of the apoptosome requires ATP or dATP [41]. It was thus argued that the switch from apoptosis to necrosis observed in Jurkat cells was due to the inability to activate the apoptosome. However, levels of dATP inside cells are usually high enough to allow apoptosome formation. Furthermore, in recent years it has been shown that ATP inhibits -rather than promote- apoptosome formation [42, 43]. Moreover, in several cell lines, reduction of ATP levels by glucose removal does not switch the form of death to necrosis, but it enhances apoptosis. Apoptosis induced by TRAIL, TNF or Fas ligation occurs faster when ATP levels are reduced by incubating cells in the absence of glucose [44, 45]. Moreover, ATP levels decrease during apoptosis, and it was shown that when mitochondrial ATP production is maintained during the apoptotic execution phase, cell membrane permeabilization is delayed [46]. Thus, at presence, there is not enough evidence to conclude that apoptosis is an energy dependent process.
DECIDING WHETHER TO DIE OR TO LIVE DEPENDING ON FOOD AVAILABILITY
Despite the common belief that ATP is required for apoptosis, most reports indicate that impairment of glycolysis generates a stress that would sensitize cells to other death inducers. Glucose deprivation or treatment with glycolytic inhibitors would thus synergize with chemotherapy, a phenomenon that can be exploited for therapeutic purposes. For instance, glycolytic inhibitors reverse resistance of leukemia cells to glucocorticoids [48] and sensitize cells from solid tumors to cisplatin, adriamycin and paclitaxel [49, 50]. The mechanism behind these effects is still unknown. It is possible that metabolic stress, like other forms of stress, promotes an imbalance in the amount of pro-apoptotic versus anti-apoptotic members of the Bcl-2 family. As discussed, nutrient levels can affect the levels or activity of p53, PUMA, Bad and Bim, which may be responsible for the sensitization or ‘synergistic’ effects of glucose deprivation with other apoptotic inducers (Fig. 2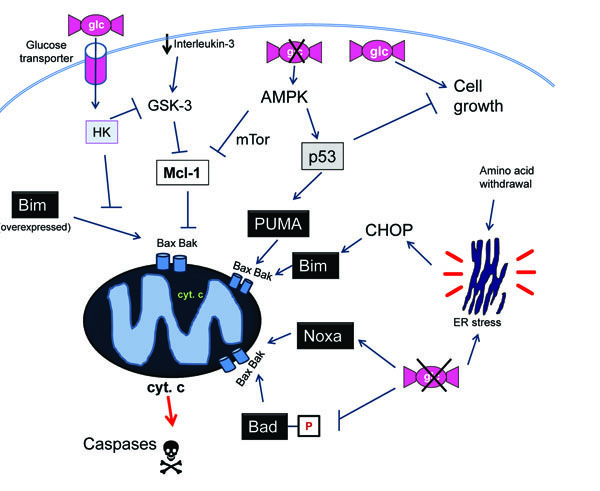 ). Moreover, it was described that glucose deprivation and 2-deoxyglucose downregulate Mcl-1 protein levels [21, 51]. In a recent report, Ricci and colleagues showed that Mcl-1 levels are reduced through an AMPK- and mTor-mediated block in translation upon inhibition of glycolysis. This makes cells susceptible to death ligands [51]. Indeed, a synergism between glycolytic inhibitors and death ligands TRAIL, FasL and TNF has been observed in several systems [44, 52, 53]. It is possible that in many cell lines glucose regulates death-ligand induced apoptosis at other levels besides Bcl-2 proteins. Sensitization to death ligands was shown to occur in some cell lines through glucose withdrawal-triggered downregulation of FLIP levels [45]. Moreover, glucose deprivation also conferred sensitivity to FasL to cells that do not require mitochondrial permeabilization to die (type I cells), and it enhanced the association of caspase-8 with TRAIL Receptor [44].
). Moreover, it was described that glucose deprivation and 2-deoxyglucose downregulate Mcl-1 protein levels [21, 51]. In a recent report, Ricci and colleagues showed that Mcl-1 levels are reduced through an AMPK- and mTor-mediated block in translation upon inhibition of glycolysis. This makes cells susceptible to death ligands [51]. Indeed, a synergism between glycolytic inhibitors and death ligands TRAIL, FasL and TNF has been observed in several systems [44, 52, 53]. It is possible that in many cell lines glucose regulates death-ligand induced apoptosis at other levels besides Bcl-2 proteins. Sensitization to death ligands was shown to occur in some cell lines through glucose withdrawal-triggered downregulation of FLIP levels [45]. Moreover, glucose deprivation also conferred sensitivity to FasL to cells that do not require mitochondrial permeabilization to die (type I cells), and it enhanced the association of caspase-8 with TRAIL Receptor [44].

These results indicate that lack of nutrients sensitizes cells to apoptosis. A slightly different interpretation of this phenomenon is that an active glucose metabolism generates a signal that protects cells from apoptosis. Could this then mean that one of the consequences of having an active glycolytic metabolism is to generate a signal that protects from death? Could an enforcement of glycolysis help cells avoid apoptosis? An elegant approach was used by Rathmell and colleagues to demonstrate that the upregulation of glycolysis per se is sufficient to confer resistance to cytokine deprivation [54]. Overexpression of a glucose transporter and a hexokinase (the enzyme that catalyzes the first step of glycolysis) was sufficient to prevent death of IL-3-dependent cells when they were deprived of this cytokine (Fig. 2). Using this same system they showed that upregulation of glycolysis prevented cytochrome c release through mainte-nance of levels of Mcl-1. Mcl-1 is normally downregulated after IL-3 deprivation due to its phosphorylation by GSK-3 (glycogensynthase kinase 3), which is a kinase that triggers Mcl-1 degradation when active [55]. Zhao et al. [56] observed that the enforcement of glucose metabolism keeps GSK-3α and 3β inactive, thus preventing the loss of Mcl-1. Interestingly, they also showed that glucose metabolism protected from the overexpression of Bim after transfection. Thus, a higher glycolytic metabolism is sufficient to confer resistance not only to growth factor removal, but also to the expression of a ‘killer’ BH3-only protein.
Another manner in which glucose metabolism impinges on apoptosis is through regulation of induction of PUMA. As we have discussed above, glucose deprivation induces PUMA. Conversely, enforcement of glucose uptake and metabolism was shown to prevent PUMA induction by growth factor withdrawal [21]. Thus, an active glycolytic metabolism may be part of the strategy that tumor cells use in order to escape social controls and survive in the absence of growth factors.
Intriguingly, nutrient deprivation can inhibit apoptosis induced by certain stimuli. For instance, apoptosis induced by some DNA-damaging chemotherapeutic agents can be reduced in the absence of glucose, or when cells are treated with 2-deoxyglucose [57, 58]. It is unclear why inhibition of glycolysis would impair cell death induced by DNA damaging agents. It is tempting to speculate that the reason is that nutrient deprivation generally causes cell cycle arrest. Since many chemotherapeutic drugs require cells to be proliferating in order to kill them, the use of an antiglycoly-tic agent which induces cell cycle arrest may be counter-productive.
TARGETING GLUCOSE AND GLUTAMINE METABOLISM FOR CANCER THERAPY
One of the hallmarks of cancer is the acquisition of resistance to apoptosis. This resistance is achieved through mutations or deregulation of levels of multiple proteins of the mitochondrial and the extrinsic pathway. Despite this, we use chemotherapy to treat tumors. Chemotherapy works because it kills proliferating cells better than non-dividing cells, and because chemotherapeutic drugs can kill cells in a non-apoptotic manner.
Interestingly, 2-deoxyglucose and other inhibitors of glycolysis are stimuli which can kill tumor cells better than non transformed cells [1]. 2-deoxyglucose is now being tested for use as a single agent or in combination as an anti-cancer agent in the United States [59] and has shown efficacy in India as a radiosensitizer [60]. The rationale behind the use of an anti-glycolitic agent for tumor treatment was provided by Warburg many years ago who showed that tumor cells have a different metabolism to normal cells. A consequence of this is that transformed cells are more sensitive to deprivation of some nutrients; particularly, glucose [1]. It was thought for many years that tumors are more glycolytic than non transformed cells because of mutations in mitochondrial DNA, which would lead them to rely on anaerobic glycolysis instead of respiration. Indeed, these mutations are frequent in tumors. Another possibility is that tumor cells have “learnt” to live without oxygen, and thus use glycolysis instead of respiration because they have to grow under low oxygen conditions. However, recently it has been realised that many of the genes that initiate tumorigenesis (oncogenes and tumor suppressors) are closely linked to metabolic regulation. p53 deficiency, transformation with c-myc or Ras, or alterations in PTEN/Akt pathway can all alter glucose and glutamine metabolism allowing cells to proliferate faster [4]. Alterations in metabolism are thought to allow tumors to avidly take nutrients and use them as building blocks for proliferation. Glucose, for instance, could mainly be used to produce nucleotides, instead of as an ATP source.
But why do tumor cells die when deprived of nutrients? This could be a similar scenario to standard chemotherapy. For example, tumor cells are more sensitive to inhibitors of DNA synthesis because they need to make more DNA than non-transformed cells. Extending this line of reasoning we could say that tumor cells are more sensitive to nutrient deprivation because they need more nutrients than non-proliferating cells. But still, why do these cells not stop cycling instead of dying? In other words, what makes a cell try to commit suicide instead of arrest and wait for better times? Although we are still far from an answer, it is possible that the reason why these cells die may be related with the inability to undergo autophagy and/or to stop the cell cycle, which occurs frequently in tumors.
One factor that can determine whether a cell dies or lives in response to nutrient deprivation is whether autophagy is engaged. As we have discussed, AMPK, the major energy sensor, is involved in cell cycle arrest and adaptation to stimuli which produce a drop in ATP levels. The signalling pathway downstream of AMPK protects cells from undergoing apoptosis in response to glucose deprivation [61]. One of the consequences of AMPK activation is induction of autophagy to help generate energy and building blocks (Fig. 1). Since tumor cells frequently display a reduced capacity to undergo autophagy, this may contribute to their susceptibility to nutrient deprivation [32].
Other downstream targets of AMPK are cell cycle inhibitors p27 and p53, which are activated by AMPK and glucose deprivation. Both of these proteins have been reported to protect cells from nutrient deprivation downstream of AMPK, an effect that may be related to the ability of these proteins to stop cell cycle but also to induce autophagy [5, 62-64]. We are hereby presented with a situation in which p53, the “bona-fide” apoptotic inducer, instead of being pro-apoptotic, protects from apoptosis. In conclusion, targeting tumor metabolism may be doubly selective for tumor cells, since it would affect proliferating cells more than quiescent cells, and on the other hand, cells that lack p53, which are frequently resistant to chemotherapy, would be more sensitive than p53-positive cells.
CONCLUSIONS AND FUTURE PERSPECTIVES
Starvation can induce cell death, but it also regulates death induced by other inducers. Solid tumors are a mass of rapidly growing cells that frequently have an imperfect blood supply and many cells within the mass may be starving or close to starvation. The high energy needs and special metabolism of tumors provide a potential that the cells natural responses to starvation can be used to target tumor cells and potentially synergise with other therapies. Therefore there is a great interest in trying to understand how cells respond to starvation and how this might interfere with existing tumor therapies. However, most researchers culture their (mostly transformed) cells in rich medium with plenty of nutrients thereby limiting their ability to detect processes that can occur in vivo, where cells compete with each other for nutrients. As an example, Alves et al. observed upregulation of Noxa by TCR stimulation, but a role for this protein on cell survival could only be detected when cells were grown in low glucose [28].
A number of questions concerning relations between cell metabolism and cell death remain to be solved. Since nutrients regulate Bcl-2 proteins, inhibitors of metabolism could synergize, positively or negatively, with other therapies. But, which apoptotic stimuli, especially the ones of clinical relevance, are more effective when combined with inhibitors of metabolism? Which BH3-only or anti-apoptotic Bcl-2 homologs are responsible for the synergistic effects observed in vitro between glucose deprivation and chemotherapy? Can this synergy be proven in animal models? Regarding the use of inhibitors of glycolysis and glutaminolysis, it remains to be determined which BH3-only proteins are responsible for death induced by inhibitors of these routes in tumor cells. Mutations in these proteins should thus predict sensitivity to anti-metabolic drugs. Moreover, could nutrient deprivation lead to autophagy-dependent necrotic cell death –since nutrient deprivation is a potent autophagic inducer- or to caspase-independent cell death, as observed in cardiomyocytes under ischemia? What are the differences, in terms of cell death, of targeting glucose metabolism, glutamine metabolism, oxygen availability or all of them? Answers to these questions may help us protect cells during ischemia, and may help us design better, more selective therapies against tumors.
NOTES
1 Ditzel M. A prickly subject: apoptotic regulation by hedgehog morphogens. Open Cell Signal J 2011; 3: 13-23.
2 Gentle I, Nachbur U. Scorched earth or viral birth? Open Cell Signal J 2011; 3: 45-51.
3 Kantari C, Walczak H. Dual philosophy in death receptor signalling. Open Cell Signal J 2011; 3: 37-44.
4 Dewson G. Interplay of Bcl-2 proteins decides the life or death fate. Open Cell Signal J 2011; 3: 31-6.
5 Ekert PG, Jabbour AM. Cytokine deprivation and cell death. Open Cell Signal J 2011; 3: 24-30.
ACKNOWLEDGEMENTS
We would like to thank J. Silke, D. Walter, S. Ramírez-Peinado and N. El Mjiyad for critical reading of this manuscript, and Isabel Fabregat and Joan Gil for support. Research in our group is supported by AICR grant 08-0621 and by grants PI071027 and RTICC RD06/0020 from the Fondo de Investigaciones Sanitarias-ISCIII.
REFERENCES
Endorsements
Table of Contents
- NUTRIENT DEPRIVATION IN PHYSIOLOGICAL AND PATHOLOGICAL SITUATIONS
- NUTRIENT DEPRIVATION IS SENSED BY AMPK AND mTOR AND LEADS TO CELL CYCLE ARREST, INHIBITION OF PROTEIN SYNTHESIS AND AUTOPHAGY
- LACK OF NUTRIENTS REGULATES BCL-2 PRO-TEINS
- NUTRIENT DEPRIVATION ALSO TRIGGERS NEC-ROSIS AND CASPASE-8 DEPENDENT APOPTOSIS
- DECIDING WHETHER TO DIE OR TO LIVE DEPENDING ON FOOD AVAILABILITY
- TARGETING GLUCOSE AND GLUTAMINE METABOLISM FOR CANCER THERAPY
- CONCLUSIONS AND FUTURE PERSPECTIVES