- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Drug Metabolism Journal
(Discontinued)
ISSN: 1874-0731 ― Volume 5, 2011
Oxidative Metabolism of Epothilones A and B (Patupilone) by Cytochromes P450: Involvement of CYP3A and CYP2C
Marie Louise Brezniceanu1, Alain Deroussent2, Helen Gu3, James B Mangold3, Hilmar Schiller4, Gerhard Gross4, Thierry Cresteil*, 1
Abstract
Epothilones are natural macrolides displaying potent antiproliferative properties against various cell lines and capable to bind tubulin and acting as microtubule-stabilizing agents like taxoids. We intended to isolate and characterize epothilone metabolites and identify enzymes implicated in the biotransformation process. In the presence of NADPH, liver microsomes from phenobarbital-treated rats produce two metabolites resulting from the oxidation of either epothilone A or B by CYP isoforms. Similarly, the oxidative biotransformation of epothilones A and B by human liver microsomes generates three metabolites with Km values ranged from 61 to 86µM. The two major metabolites (m1 and m2) are hydroxylated on the macrolide ring essentially by CYP3A4, whereas 3A5, 3A7 and 2B6 are minor contributors to the reaction. M3 is formed by CYP2C19 and 2C9 and results from the hydroxylation of the methyl on carbon 17 of the lateral chain. Inhibition of CYP3A almost completely abolished the formation of m1 and m2, whereas inhibition of CYP2C19 substantially reduced the production of m3. Collectively these data suggest that the oxidative metabolism of epothilones is principally mediated by CYP3A4 and CYP2C19. Epothilone B was found to be an in vitro inhibitor of CYP2C9 [IC50≈25µM], CYP2C19 [Ki≈1.7µM] and CYP3A4/5 [Ki≈1.85µM] whereas conversely taxanes or Vinca alkaloids significantly reduced oxidation of epothilone B. However, clinically relevant inhibition in patients undergoing chemotherapy is unlikely due to low therapeutic epothilone B blood concentrations.
Article Information
Identifiers and Pagination:
Year: 2008Volume: 2
First Page: 14
Last Page: 23
Publisher Id: TODMJ-2-14
DOI: 10.2174/1874073100802010014
Article History:
Received Date: 25/3/2008Revision Received Date: 14/10/2008
Acceptance Date: 15/10/2008
Electronic publication date: 24/12/2008
Collection year: 2011
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the ICSN, Avenue de la terrasse, 91198 Gif sur Yvette, France; Tel/Fax: 33 169 823 643;E-mail: cresteil@icsn.cnrs-gif.fr
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 25-3-2008 |
Original Manuscript | Oxidative Metabolism of Epothilones A and B (Patupilone) by Cytochromes P450: Involvement of CYP3A and CYP2C | |
INTRODUCTION
Epothilones (Epo) are natural products isolated from the myxobacteria Sorangium cellulosum and were initially selected for their antifungal properties [1]. They are 16-membered macrolides whose structure has been elucidated by Höfle et al. [2], being epothilone B the 12-methyl derivative of epothilone A. Along with their antifungal properties, Epo displayed potent cytotoxic activity against the mouse fibroblast L929 line (IC50 of 15 and 2 ng/ml, respectively for Epo A and Epo B, approximately 30 and 4 nM) and the human T-24 bladder carcinoma cell line with an IC50 value of 50 nM for Epo A [1]. Epo B (patupilone, EPO906) has been in clinical development for treatment of solid tumors [3] as well as other epothilone analogs (BMS247550, BMS310705, Fludelone, Ixabepilone) but so far no available data are been published in the literature regarding preclinical or clinical pharmacokinetic or metabolism of epothilone B alone or in combination in human cancer patients. Independently, Bollag and colleagues [4] identified Epo as microtubule-stabilizing agents capable to competitively inhibit the binding of paclitaxel to tubulin. Basically, the biological effects of Epo mimicked those of paclitaxel in inducing in vitro tubulin polymerization into microtubules and enhanced their stability and bundling in cultured cells. The result was a cell cycle arrest in phase G2/M leading to cytotoxicity. Epo B however exhibits a more potent binding and a higher affinity to (-tubulin compared to paclitaxel [5, 6] and the binding sites are probably different [7]. In addition, Epo B is capable of binding to mutated (-tubulin [8]. Importantly, the ability of Epo A and B to elicit mitotic arrest and cytotoxicity was only moderately affected by the hyperexpression of p-glycoprotein in cultured cell lines, in contrast with the high resistance to paclitaxel acquired by cell lines [4, 5, 9]. When given in vivo, Epo B displayed a stronger cell growth inhibition compared to paclitaxel [9, 10-12].
Similarities existing in the mode of action of paclitaxel and Epo might be explained by some structural analogies: as noticed by Höfle et al. [2] both compounds contain a small ether ring, a geminal dimethyl group and a (-hydroxyoxo unit. In addition the size of the taxane ring approximatively corresponds to the size of the macrolide ring of Epo. Recent efforts to superimpose the structures of Epo and paclitaxel have been published in order to define a common pharmacophore motif able to bind tubulin [7, 8, 13]. However, there are also clear structural differences between epothilones and taxoids like the lactone group of Epo, which gives rise to a different mode of metabolic inactivation due to hydrolysis [14].
The pharmacological effect of antimitotic agents at the cellular level depends on their concentration and consequently their biotransformation and elimination. Due to the complexity of its structure, paclitaxel could undergo several metabolic pathways: thus at the hepatic level, paclitaxel is subject to hydroxylation by cytochromes P450 before being excreted via the bile route in an unconjugated form. In the human, the major metabolite formed results from the hydroxylation on carbon 6 of the taxane ring by CYP2C8, whereas a minor metabolite corresponds to the hydroxylation of the phenyl ring on the lateral chain catalyzed by CYP3A4 [15-17]. In case of docetaxel, the hydroxylation on the tert-butyl group of the lateral chain was the only reaction occurring in human liver microsomes, this reaction being clearly assigned to CYP3A4 and 3A5. Interestingly, docetaxel was capable to completely inhibit the hydroxylation on the lateral chain of paclitaxel, but had no effect on the 6(-hydroxylation of the taxane ring [18].
Thus it becomes evident that the hydroxylation of taxoids by CYP protein is regioselective, probably in relation with the insertion of substrates into the active sites of CYP3A4 and CYP2C8. Assuming a partial structural homology between taxoids and epothilones, we were interested in the elucidation of the oxidative metabolism of epothilones in order to characterize the metabolites generated during in vitro incubations with rat and human liver microsomes and to identify the CYP proteins involved in these reactions. The ability of Epo B to inhibit monooxygenase activities was investigated in pooled human liver microsomes and finally interactions between Epo B and drugs used in chemotherapy were investigated in vitro.
MATERIALS AND METHODOLOGIES
Chemicals
Epo A and B (Epo B, EPO906) were provided by Novartis Pharma [Basel, Switzerland)(purity ≥98% as shown in HPLC), SKF525A by SmithKline Beecham (Brentford, UK), docetaxel and iritotecan (CPT11) by Rhone Poulenc Rorer (Vitry, France), paclitaxel by Bristol Myers Squibb (Wallingford, CT). Sulfaphenazole and anti-CYP3A4 inhibition kit were purchased from Gentest (Woburn, MA), diclofenac, phenacetin, chlorzoxazone, testosterone, midazolam, trichloropropane oxide, erythromycin, ketoconazole, furafylline, ciclosporin A, etoposide, vincristine, vinblastine, verapamil and troleandomycin were obtained from Sigma-Aldrich (St Quentin-Fallavier, France) and ticlopidine from ICN Pharmaceuticals (Orsay, France). HPLC columns were purchased from Interchim (Montluçon, France) and Phenomenex (Torrence, CA).
Tissue Collection
Liver samples were obtained from adult donors for organ transplantation after approval by the Ethical Committee of the Institut National de la Santé et de la Recherche Médicale. Donors had no drug history and died from traffic accident. No information was available about their smoking habits. Samples were removed within minutes after circulation arrest, immediately frozen into liquid nitrogen and kept at -80°C until use.
Microsome Preparation and Determination of Individual Protein Content
Male Sprague Dawley rats (200g) were treated i.p. with phenobarbital in saline (80mg/kg) daily for 3 days. After sacrifice, livers were excised and microsomes prepared as reported earlier [19]. Frozen human livers were thawed into ice-cold isotonic saline, homogenized and processed as rat livers. Microsomes were resuspended into 100 mM sodium phosphate buffer, pH 7.4, 20% glycerol: their cytochrome P450 content was estimated by the procedure of Greim [20] and the protein concentration was measured by a micro BCA kit (Pierce). Monooxygenase activities and the presence of individual CYP proteins were determined in liver microsomes as stated previously [21-24].
CYP Expressing Cell Lines
Full length cDNAs coding for human CYP1A1, 1A2, 2A6, 2C8, 2C9, 2C18, 2C19, 3A4, 3A5 and 3A7 were inserted in the correct orientation of the expression vector pMT2 as previously reported [22-24]. Ad 293 cells were cotransfected with 50µg of CYP-pMT2 plasmid and 5µg of vector conferring the resistance to geneticin. Full length cDNAs for CYP2B6, 2D6 and 2E1 were cloned in pCIneo (Promega, Madison, WI). Clones growing in presence of G418 and expressing a high level of CYP protein determined by immunoblotting were selected as previously described [22-24]. In case of CYP3A4, 3A5 and 3A7, stable cell lines were further transfected with the full length cDNA encoding human NADPH-cytochrome P450 reductase inserted into pMT2. The addition of multiple copies of the reductase gene markedly increased the production and activity of the reductase protein and consequently stimulated monooxygenase activities supported by CYP3A genes but had no action on the catalytic activity of other CYP proteins. Cells near half-confluence in 75 cm2 flasks were incubated for 24h with 50µM Epo in 4 ml Dulbecco-MEM medium. Epo and their metabolites were extracted three times with 5 ml dichloromethane and processed as indicated below.
Oxidative Metabolism of Epo A and B
Rat liver microsomes, corresponding to 1 nmole total P450, were incubated in a final volume of 0.5 ml of 50 mM sodium phosphate buffer pH 7.4, 5 mM MgCl2, 10% glycerol containing 0.1 mM NADP and 1 mM glucose 6 phosphate. Incubation conditions were selected after optimization for the production of metabolites. Epo A and B dissolved in methanol to give a stock solution at 10 mM, were added to a final concentration of 50 µM (except when otherwise indicated) and the reaction was initiated by the addition of glucose-6-phosphate dehydrogenase. The incubation was carried out at 37°C for 30 minutes and was stopped by the addition of 2 ml dichloromethane. Extraction was repeated twice: organic phases were pooled and evaporated to dryness under nitrogen.
Human liver microsomes (1 mg protein) were similarly processed except that incubations were carried out for 60 minutes. Kinetic parameters were measured in incubations performed following the same protocol with increasing concentrations of Epo ranging from 0 to 100 µM. For inhibition studies, chemicals dissolved in a minimum volume of methanol were added to the incubation mixture in the presence of the NADPH generating system for a 10 min preincubation and the reaction was initiated by the addition of Epo. Immunoinhibition studies with CYP3A4 antibodies were performed following the recommendation of the manufacturer.
Characterization and Quantitation of Epothilone Metabolites
Dry residues were dissolved in methanol and analyzed by HPLC. Briefly, aliquots were injected onto a Zorbax C18 column (250x4.6 mm, 5µm) fitted with a guard column. Elution was performed at a flow rate of 1 ml/min with a mixture of 60% methanol, 40% water and was monitored at 254 nm. Under these conditions, retention times for Epo A and B were 14.9 and 18.3 minutes, respectively. For identification of Epo metabolites in LC/MS/MS analysis, samples were injected in the HP1100 model HPLC system (Agilent Technologies, Massy, France) including a binary pump and a by-pass loop autosampler and a microbore C18 Nucleosil column (50 x 1 mm, 5µm) (Interchim, France). The elution was realized at 50µl/min with a gradient of acetonitrile from 20 to 80% in water-0.1%formic acid. The column outlet was connected in line to a UV detector set at 254 nm and the orthogonal electrospray source of a triple quadrupole mass spectrometer (Quattro-LCZ, Micromass, Manchester, UK) operated in precursor ion scan and in product ion scan mode with argon as a collision gas. The collision energy was set at 30V. Positive ion electrospray spectra were recorded over the mass range m/z 80 to 600 in 1 sec. The spray voltage was set at 3500V and the cone voltage of the ion source was kept at 40V.
Inhibition of Monooxygenase Activities by Epo B
For analysis of monooxygenase inhibition, pooled human liver microsomes (lot no 0110186, total CYP content of 0.57 nmoles.mg-1 protein) were purchased from XenoTech (Lenexa, KS). Assays were performed at 37°C in a final volume of 100µl containing 100 mM phosphate buffer (pH 7.4), 5 mM MgCl2 and 0.2 or 0.5 mg/ml microsomes. Probe substrates dissolved in DMSO (0.2% final concentration) were incubated at the concentrations stated in Table 2. Epo B was added in a concentration range between 1 to 100 µM. Reactions were initiated by the addition of 1 mM NADPH after a 3 min preincubation period and terminated after 10-30 min by addition of 200µl acetonitrile. Samples were evaporated and reconstituted in 100 µl 5/95 (v/v) acetonitrile/water. After addition of internal standard warfarin (measurement in positive ion mode) or 4’-hydroxy-S-mephenytoin (negative ion mode), 10 µl aliquots were analyzed by LC/MS/MS with a Shimadzu liquid chromatograph equipped with a PE Sciex API 3000 (Applied Biosystems, Foster City, CA) mass spectrometer. Chromatographic separation was achieved with a Phenomenex polar RP column (50x2.1mm, 4µm). The mass spectrometer was operated in multiple reaction monitoring (MRM) mode with the Turbo Ion Spray probe at 380°C.
RESULTS
Metabolism of Epo A by Rat and Human Liver Microsomes
When the incubation mixture of Epo A with liver microsomes isolated from PB-treated rats was subjected to HPLC, two peaks called m1 and m2 eluting at 6.9 and 7.8 min were monitored at 254 nm in addition to the parent molecule which had a retention time of 14.3 min (data not shown). When NADPH was omitted from the incubation, the formation of these products was severely impaired suggesting the participation of CYP proteins. To definitively demonstrate their involvement in the reaction, inhibitors were used to prevent the formation of the derivatives. Trichloropropane oxide (TCPO) selectively inhibits microsomal epoxide hydrolase [25]: when added to the incubation, TCPO failed to reduce the production of Epo A derivatives. Furthermore when Epo A was incubated in the absence of NADPH with microsomal preparations or cytosol, no diol resulting from the hydration of the epoxide by microsomal or cytosolic epoxide hydrolase was formed. This suggested that under these conditions the epoxide was quite stable and that epoxide hydrolase did not play a role in the in vitro biotransformation of Epo A. Conversely, SKF 525A a non-specific inhibitor of CYP, almost completely prevented the formation of the derivatives thus involving CYP proteins.
Structure of Epothilone A Metabolites
When incubated with 50µM Epo A, human liver microsomes formed metabolites eluting with the same retention time as the metabolites generated by rat microsomes. The structure of Epo A and its metabolites was elucidated by LC/MS/MS. The mass spectrum of authentic Epo A showed fragment ions at m/z 406, 306, 166 and 113 in addition to the molecular mass ion at (M+H+) 494. These fragments resulted from the release of the lateral chain (m/z 166 and 113) or from the breakage of the macrolide ring (m/z 406 and 306). Similarly analyzed, the Epo A derivatives m1 and m2 yielded the same molecular mass of 509 [(M+H+) 510]: this increase of 16 of the molecular mass would indicate the addition of an hydroxyl group on the Epo A molecule. This was further evidenced by the loss of two molecules of water (m/z 492 and 474) consistent with the presence of two hydroxyl groups, whereas only one group could be released from the native Epo A molecule (m/z 476). The fragmentation of metabolites m1 and m2 generated fragments at m/z 322 and m/z 422 indicating that the additional hydroxyl group was inserted on the macrolide ring, whereas the lateral chain remained unchanged with fragments at m/z 166. Taken together these data clearly suggest that the hydroxylation of Epo A occurs on the macrolide ring, most likely on carbons 9, 10 or 11.
The rate of oxidative biotransformation of Epo A is related to the CYP content of microsomes: in rat 1 nmole of total P450 hydroxylated roughly 16 nmoles of Epo A in 30 minutes, while 8-9 nmoles of Epo A were transformed by 0.3 nmoles of human P450 in 1 hour. Thus, the turnover number or molecular activity of the monooxygenase system towards Epo A was comparable in rat and human when expressed as nmole metabolite formed by 1 nmole total P450.
Oxidative Metabolism of Epothilone B and Structure of its Metabolites
When 50 µM Epo B were incubated with human liver microsomes under the same conditions as Epo A, two peaks called m1 and m2 were eluted by HPLC with a retention time of 7.8 and 9.1 min versus 18.3 min for the parent Epo B molecule (data not shown). Examination by LC/MS/MS showed Epo B to produce a molecular mass of 507 [(M+H+) 508] with fragments at m/z 420, 320, 166 and 113 (Fig. 1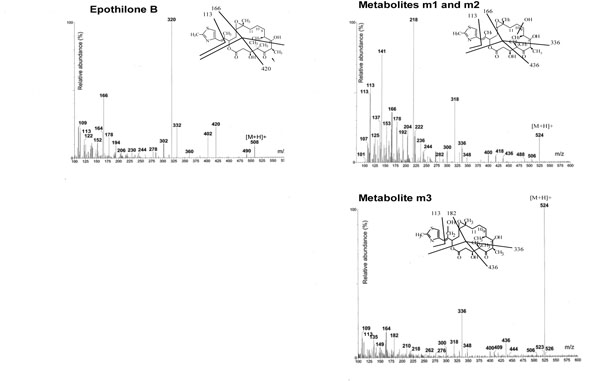 ). Its metabolites yielded a (M+H+) ion at 524 indicating a monohydroxylation of the parent molecule and the pattern of fragmentation strongly suggested that the hydroxylation site was located on the macrolide ring with fragments at m/z 436, 336, 166 and 113 (Fig. 1). Thus hydroxylation of Epo B catalyzed by human liver microsomes appeared to be quite similar to that of Epo A.
). Its metabolites yielded a (M+H+) ion at 524 indicating a monohydroxylation of the parent molecule and the pattern of fragmentation strongly suggested that the hydroxylation site was located on the macrolide ring with fragments at m/z 436, 336, 166 and 113 (Fig. 1). Thus hydroxylation of Epo B catalyzed by human liver microsomes appeared to be quite similar to that of Epo A.
 |
Fig. (1) Mass spectra of Epo B and its metabolites generated by human liver microsomes. Inserts are the proposed fragmentation patterns. |
To identify the hydroxylated products of Epo A and B, we also used cultured cell lines expressing individual recombinant CYP proteins. In addition to metabolites m1 and m2 already described, a new metabolite called m3 was isolated from incubations of Epo A and B with CYP2C19 or 2C9 (Fig. 2A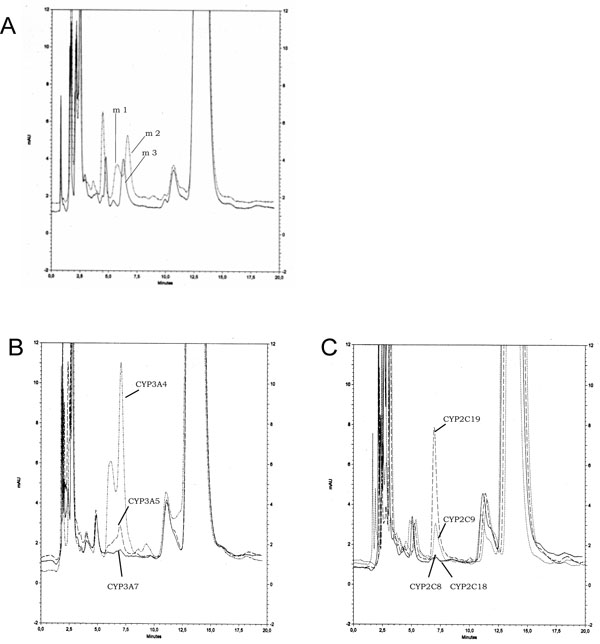 ). Examination by LC/MS/MS of Epo A m3 and Epo B m3 showed that these metabolites resulted from the monohydroxylation of the parent molecules with an m/z at +16 as for m1 and m2. However the fragmentation pattern appeared different with a fragment at 182 instead of 166 suggesting that the reaction occurred on the lateral chain but not on the thiazole ring with a conserved fragment at m/z 113. The proposed structure of Epo B m3 is shown in Fig. (1).
). Examination by LC/MS/MS of Epo A m3 and Epo B m3 showed that these metabolites resulted from the monohydroxylation of the parent molecules with an m/z at +16 as for m1 and m2. However the fragmentation pattern appeared different with a fragment at 182 instead of 166 suggesting that the reaction occurred on the lateral chain but not on the thiazole ring with a conserved fragment at m/z 113. The proposed structure of Epo B m3 is shown in Fig. (1).

Kinetics of the Oxidative Biotransformation of Epo A and B
The kinetic parameters of Epo A and B hydroxylation were calculated using rat and human liver microsomes. Km values for Epo A calculated individually in three different human livers ranged from 19 to 38 µM and were 2- to 4-times lower than in microsomes isolated from a pool of PB-treated rats (77µM). The higher P450 content of rat liver microsomes (2.8 nmoles P450/mg) as compared with human liver microsomes (0.38 nmol/mg) accounts for the important difference in the maximal velocity of Epo A transformation existing between rat and human preparations (0.19±0.11 nmol.min-1.mg-1 vs 3.8 in rats). Km for Epo B were higher than for Epo A in human samples (74±12µM), but maximal velocities were in the same range (0.24±0.14 nmol.min-1. mg-1) suggesting that the affinity of human liver microsomes was slightly lower for Epo B than for Epo A, with identical catalytic efficiencies.
When the oxidative biotransformation of 50 µM Epo A or B was estimated in 16 human liver microsomes previously screened for their monooxygenase activities, a wide interindividual variability in the hydroxylation rates by human liver microsomes was noticed: the formation of metabolites was 31.2±26.5, 84.5±60.6 and 13.3±7.0 pmoles.min-1.mg-1 respectively for m1, m2 and m3. The formation of Epo B m1 was highly correlated with the formation of Epo B m2 (r=0.958) but less correlated with the formation of m3 (r=0.862). When compared with the isoform content of microsomes, a positive correlation was noted between the formation of Epo B m2 and the CYP3A content (r=0.86) and its testosterone 6β hydroxylase activity (r=0.74), but not with the CYP2C content (r=0.56). When the biotransformation of taxoids was examined in the same hepatic samples, the correlation existing between the formation of Epo B m2 and CYP 3A dependent activities was high: r=0.873 and 0.733 respectively with the hydroxylation on the lateral chain of docetaxel and paclitaxel, whereas the hydroxylation on the taxane ring of paclitaxel supported by CYP2C8 was only poorly correlated (r=0.45). Thus it appears that the formation of metabolites m1 and m2 by human liver microsomes is predominant and could be attributed to CYP3A isoforms whereas metabolite m3 produced by CYP2C is of minor importance.
Identification of CYP Involved in Epothilone Metabolism
To tentatively identify the CYP protein(s) involved in the hydroxylation of Epo A and B, several approaches have been combined. In a first attempt, we used CYP specific inhibitors in three individual human liver microsomal preparations. The formation of individual metabolites m1, m2 and m3 of Epo B was examined and displayed in Table 1: 2µM ketoconazole (inhibitor of CYP3A [26] was a potent inhibitor of the formation of m1 and m2 with 83-100% inhibition, whereas ticlopidine (CYP2C19 inhibitor, [27]) reduced by 50% the production of m3 but has no effect on m1 and m2. Sulfaphenazole (a selective inhibitor of CYP2C9 [26]) and furafylline (CYP1A2 [28]) had no or weak inhibitory effect (<10% inhibition) on either metabolites.

Then inhibition of Epo B metabolism by immunoglobulins raised against CYP3A4 was tested in human liver microsomes. Increasing amounts of serum containing anti-CYP3A4 immunoglobulins inhibited the hydroxylation of Epo B in a dose-dependent manner to reach more than 90% inhibition (data not shown). This clearly confirmed the role of CYP3A isoforms in the oxidative metabolism of Epo.
To definitively ascertain the involvement of CYP proteins in the hydroxylation of epothilones we used cultured cell lines expressing individual recombinant CYP proteins. The formation of metabolites m1 and m2 was mainly detectable in the culture medium of Ad293 cells stably transfected with CYP3A. As in incubations with human liver microsomes the formation of metabolite m2 exceeded that of metabolite m1. Cells expressing CYP3A4 had the highest transformation rate: when incubated with 50µM Epo B for 24h, the production of m1 and m2 accounted for about 5 nmoles per mg cellular proteins in 24 h. Cell lines expressing CYP3A5 and 3A7 had lower activities (Fig. 2B) as well as those expressing CYP2A6, while CYP2B6 and 1A2 had a marginal activity (Table 2). On the other hand, metabolite m3 was mostly produced by CYP2C isoforms: cell line expressing CYP2C19 had the highest activity followed by CYP2C9 (Fig. 2C). Cell lines expressing CYP2C18 and 2C8 exhibited a marginal activity together with CYP2A6. All other cell lines expressing P450 had no detectable activity (Table 2). These metabolites were isolated and analyzed by LC/MS/MS: their fragmentation profiles were identical with those of metabolites obtained with rat and human liver microsomes and thus confirmed former structures.
Similarly Epo A incubated with Ad293 derived cell lines was hydroxylated mainly by cells expressing 3A or CYP2C19 and 2C9. The production of metabolites m1 and m2 accounted for about 15% of 50µM Epo A added to the culture medium, i.e. 30 nmoles formed in 24h per flask.
All together, these data clearly point out that CYP3A4 is the principal isoform responsible for the hydroxylation of Epo A and B, whereas CYP2C19 has a minor participation.
Inhibition of Monooxygenase Activities by Epothilone B
Epo B was evaluated for its ability to inhibit specific monooxygenase activities catalyzed by human liver microsomes. For that purpose, a pool of microsomes prepared from human livers was used and 8 monooxygenase activities representative of the major CYPs expressed in the liver were assayed in the presence of increasing concentrations of Epo B ranging from 1 to 100 µM. Concentrations eliciting 50% inhibition of the probe substrate activity in the absence of Epo B were calculated and shown in Table 3.
Two CYP3A4/5 dependent activities (testosterone 6β-hydroxylation and midazolam 1 hydroxylation) were inhibited by Epo B in a dose-dependent manner as well as the hydroxylation of S-mephenytoin by CYP2C19 (Fig. 3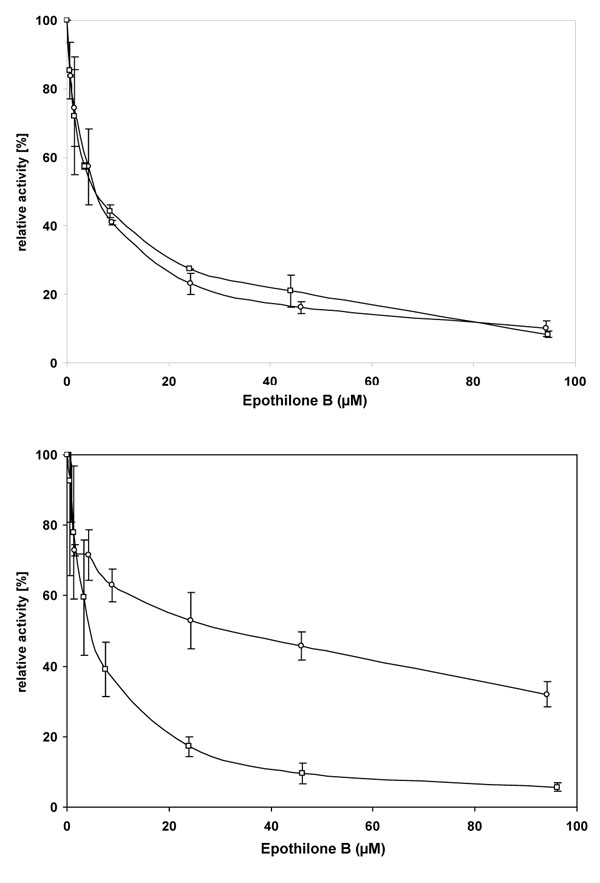 ). All three reactions were inhibited by Epo B with an IC50 ≈ 5 µM and the maximal extent of inhibition was higher than 90%. Diclofenac 4’-hydroxylation, a marker for CYP2C9 activity, was moderately inhibited (up to 70%) by Epo B with an IC50 ≈ 25µM, whereas other activities representative of CYP1A2, 2C8, 2D6 and 2E1 showed little or no inhibition by Epo B (IC50 ( 75µM). The inhibition of CYP3A4/5 and CYP2C19 by Epo B was further investigated by the determination of Ki. The 4’-hydroxylation of mephenytoin in pooled liver microsomes was competitively inhibited by Epo B with a Ki of 1.75µM in three separate experiments (data not shown). Similarly, for the 1’-hydroxylation of midazolam a Ki of 1.85µM was calculated.
). All three reactions were inhibited by Epo B with an IC50 ≈ 5 µM and the maximal extent of inhibition was higher than 90%. Diclofenac 4’-hydroxylation, a marker for CYP2C9 activity, was moderately inhibited (up to 70%) by Epo B with an IC50 ≈ 25µM, whereas other activities representative of CYP1A2, 2C8, 2D6 and 2E1 showed little or no inhibition by Epo B (IC50 ( 75µM). The inhibition of CYP3A4/5 and CYP2C19 by Epo B was further investigated by the determination of Ki. The 4’-hydroxylation of mephenytoin in pooled liver microsomes was competitively inhibited by Epo B with a Ki of 1.75µM in three separate experiments (data not shown). Similarly, for the 1’-hydroxylation of midazolam a Ki of 1.85µM was calculated.
 |
Fig. (3) Inhibition of monooxygenases in human liver microsomes by Epo B. |
Drug-Drug Interactions with Antineoplastic Molecules
To investigate the possible interaction of antineoplastic drugs with the oxidative metabolism of epothilones, we incubated Epo A and B with a pool of human liver microsomes in the presence of a series of drugs used in chemotherapy or with drugs known for their affinity for CYP3A isoforms. Concentrations of Epo A and B chosen for those experiments corresponded to twice the Km for Epo (50 and 120µM respectively for Epo A and B) and for drugs when available from the literature. Results of the competition between Epo B and drugs were shown in Fig. (4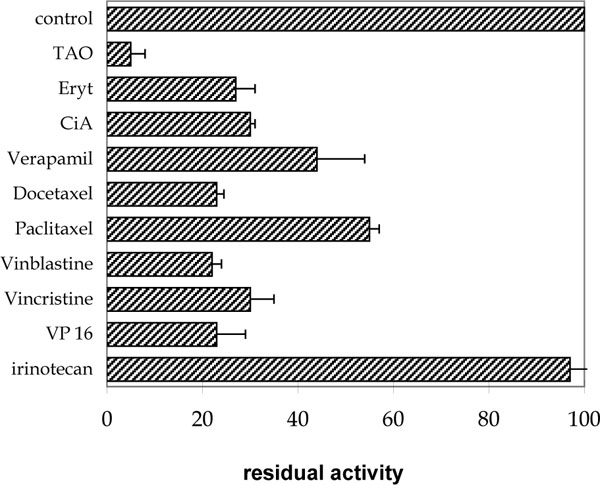 ). The most potent inhibitor of Epo B hydroxylation was troleandomycin which at a dose of 10µM reduced the formation of hydroxyepothilone B by 95 %. A similar inhibition was observed for the hydroxylation of Epo A. Erythromycin (400µM) and cyclosporin A (25µM) also actively inhibited the formation of the Epo B hydroxyl metabolites. Among antineoplastic drugs, vinblastine (15µM), vincristine (15µM), etoposide (VP16, 100µM) and docetaxel (5µM) exerted a pronounced effect on the hydroxylation of Epo B whereas paclitaxel (30µM) was less active. Irinotecan (CPT11) at a dose of 60 µM had no effect on the oxidative biotransformation of either Epo A or B.
). The most potent inhibitor of Epo B hydroxylation was troleandomycin which at a dose of 10µM reduced the formation of hydroxyepothilone B by 95 %. A similar inhibition was observed for the hydroxylation of Epo A. Erythromycin (400µM) and cyclosporin A (25µM) also actively inhibited the formation of the Epo B hydroxyl metabolites. Among antineoplastic drugs, vinblastine (15µM), vincristine (15µM), etoposide (VP16, 100µM) and docetaxel (5µM) exerted a pronounced effect on the hydroxylation of Epo B whereas paclitaxel (30µM) was less active. Irinotecan (CPT11) at a dose of 60 µM had no effect on the oxidative biotransformation of either Epo A or B.

DISCUSSION
CYP proteins display regioselective reactions towards endogenous and exogenous molecules depending on the topology, size and hydrophobicity of their substrate binding site. With paclitaxel and docetaxel, it was previously demonstrated that CYP3A4-dependent hydroxylation occurred exclusively on the lateral chain, either at the tert-butyl or the phenyl ring at C'3, whereas hydroxylation on the taxane ring was only supported by CYP2C8 [15, 17, 18]. This could be interpreted in terms of an opposite orientation of paclitaxel into the binding sites of the two proteins. Based on the proposed partial structure homology between taxoids and epothilones, it could be hypothesized that epothilones might be substrates for both CYP2C8 and 3A4, leading to the formation of hydroxylated metabolites on the lateral side-chain and its thiazole ring or on the macrolide ring of epothilones.
Data collected in the present study demonstrate that the oxidative metabolism of epothilones by rat and human liver microsomes generates three metabolites resulting from the mono hydroxylation of the native molecule by CYP proteins. The epoxide is not hydrated by either microsomal or cytosolic epoxide hydrolase from rat and human preparations and seems to be quite stable. M1 and m2 are the major oxidative metabolites formed by human liver microsomes during in vitro incubations and exhibit the same fragmentation profile. We assumed that the site of hydroxylation is located on the macrolide ring, most likely on carbons 9, 10 or 11. M1 and m2 are probably isomers but the exact site of hydroxylation has to be confirmed by nmr analysis on large scale preparations of epothilone metabolite in a next set of experiments. M3 results from the hydroxylation on the lateral chain of Epo A or B but not on its thiazole ring and is produced in lower amounts than m1 and m2. Therefore we propose that the hydroxylation site is the methyl group on C17 of the lateral chain. To date we have no information on the biological activity of these metabolites with regard to tubulin assembly and cytotoxicity in cultured cell lines and in animals. However a recent report by Tang et al. demonstrates that natural hydroxylated derivatives of Epo D are less cytotoxic on several cell lines than the native Epo D molecule [29].
The in vitro oxidative biotransformation of epothilones into three metabolites was repeatedly observed with rat and human liver microsomes as well as with human recombinant enzymes. It shows no qualitative difference in Epo A and B metabolism. The assumption that CYP3A4 is the major isoform active in the oxidation of epothilone is based on several criteria: first, the recombinant CYP3A4 protein expressed in mammalian cells is the most efficient to catalyze the hydroxylation of Epo A and B into the major oxidative metabolites m1 and m2, a reaction which could be almost completely prevented by addition of CYP3A4 inhibitors to human liver microsomes or by immunoinhibition with antibody raised against CYP3A4. In addition to CYP3A4, CYP3A5 is also active in the generation of m1 and m2 but to a much lower extent. The lower capacity of CYP3A5 to carry out the hydroxylation reaction could result from a poor affinity of epothilones for the CYP3A5 binding site. Such a difference between the affinity of CYP3A4 and 3A5 has already been reported for docetaxel and could explain that CYP3A5 participates moderately to docetaxel biotransformation [30]. Furthermore, the polymorphic expression of CYP3A5 in only 25% of the population [31] reduces its relative contribution to the overall metabolism of docetaxel or epothilones in the human liver.
Upper panel: 1-100 µM Epo B was incubated with 25µM testosterone or 5µM midazolam for 10 min with pooled human liver microsomes. The formation of 6β-hydroxytestosterone [□) and 1’-hydroxymidazolam (○) was monitored by LC/MS. Results are expressed as the percentage of residual activity measured without Epo B and represent the mean of three separate experiments.
Lower panel: 1-100 µM Epo B was incubated with 5 µM diclofenac for 10 min or 15 µM S-mephenytoin for 20 min with pooled human liver microsomes. The formation of 4(-hydroxydiclofenac (○) and 4(-hydroxy S-mephenytoin (□) was monitored by LC/MS. Results are expressed as the percentage of residual activity measured without Epo B and represent the mean±SD of three separate experiments.
Conversely the formation of the third minor oxidative metabolite m3 is essentially supported by CYP2C19 and by CYP2C9 to a lower extent. This is consistent with the inhibition of m3 formation in human liver microsomes by ticlopidine and the modest inhibition elicited by sulfaphenazole. Despite its predominant role in the 6( hydroxylation of paclitaxel, CYP2C8 has no role in the biotransformation of epothilones. This suggests that the insertion of Epo B in the active site of CYP2C8 was quite different from that of paclitaxel. This was already reported for the hydroxylation of docetaxel which was not oxidized by CYP2C8. Other P450 proteins have no or negligible roles in the biotransformation of epothilones.
This study focuses on P450 reactions. However, in vivo data collected from nude mice treated with Epo B [14] indicate that oxidative reactions represent a minor part of total Epo B metabolism. The major metabolic pathway for this compound was found to be in mice hydrolysis of the ester and epoxide functions producing inactive free acid metabolites. To date no data was available in the literature regarding the relative importance of the different metabolic pathway in the human species.
The involvement of CYP3A4 and 2C19 in oxidative metabolism of Epo B is consistent with the ability of Epo B to reduce the catalytic activities supported by CYP in human liver microsomes. Only CYPs capable to bind and oxidize Epo B, namely CYP2C19 and CYP3A4/5, were markedly inhibited by Epo B. Thus Epo B is a potent ligand for CYP3A4/5 and CYP2C19 and competitively inhibits monooxygenase activities supported by these isoforms. Furthermore, a direct relationship was reported within the CYP2C subfamily between the rate of Epo B biotransformation and the extent of inhibition estimated by the IC50 value of Epo B: CYP2C8 was inactive in the biotransformation reaction and was not inhibited, CYP2C9 was weakly inhibited and had an intermediate activity whereas CYP2C19 was the most active CYP2C enzyme and displayed the strongest inhibition of all CYP2C enzymes by Epo B. This view is consistent with competitive inhibition of monooxygenase activities by Epo B. Although Ki values measured in this study are in the low micromolar range, clinically relevant inhibition by Epo B is not likely because therapeutic Epo B blood concentrations are in the low nanomolar range [3] and therefore much lower than the inhibition constants.
A consequence of the major role of CYP3A4 in epothilone oxidation is the possibility of inhibition by other CYP3A4 substrates or inhibitors used in clinics and possibly associated with epothilones in cancer patients. Taxoids and Vinca alkaloids incubated at concentrations twice higher than their determined Km values for CYP3A4 are potent inhibitors of oxidative epothilone metabolism [15, 18, 32]. However the concentrations used in in vitro studies largely exceeded the plasma concentrations observed in vivo and should be taken with caution before any extrapolation to the clinical situation. Surprisingly, irinotecan has no effect on Epo A and B oxidative metabolism. Recently, the hydroxylation of irinotecan was attributed to CYP3A4 and to a lesser extent to CYP3A5 [33, 34] with Km values around 20-40 µM. However, under our conditions 60 µM irinotecan totally failed to impair epothilone hydroxylation. A poor solubility of irinotecan in aqueous solution lowering its real concentration in the binding site or a non-competitive inhibition might account for that unexpected observation but to date we have no satisfactory explanation.
In conclusion, the oxidative metabolism of Epo A and B is mainly supported by CYP3A4 and CYP2C19 and this metabolic pathway could be inhibited to some extent by other antineoplastic drugs.
ACKNOWLEDGEMENT
This work was supported by a grant from Association de Recherche sur le Cancer (number 5464).