- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
Fabry Disease – Current Treatment and New Drug Development
Omid Motabar1, Ellen Sidransky*, 1, Ehud Goldin1, Wei Zheng*, 2
Abstract
Fabry disease is a rare inherited lysosomal storage disorder caused by a partial or complete deficiency of α-galactosidase A (GLA), resulting in the storage of excess cellular glycosphingolipids. Enzyme replacement therapy is available for the treatment of Fabry disease, but it is a costly, intravenous treatment. Alternative therapeutic approaches, including small molecule chaperone therapy, are currently being explored. High throughput screening (HTS) technologies can be utilized to discover other small molecule compounds, including non-inhibitory chaperones, enzyme activators, molecules that reduce GLA substrate, and molecules that activate GLA gene promoters. This review outlines the current therapeutic approaches, emerging treatment strategies, and the process of drug discovery and development for Fabry disease.
Article Information
Identifiers and Pagination:
Year: 2010Volume: 4
First Page: 50
Last Page: 56
Publisher Id: CCGTM-4-50
DOI: 10.2174/1875397301004010050
Article History:
Received Date: 3/4/2010Revision Received Date: 3/6/2010
Acceptance Date: 14/6/2010
Electronic publication date: 23/7/2010
Collection year: 2010
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to these authors at the Medical Genetics Branch, National Human Genome Research Institute, National Institutes of Health, 35 Convent Drive, MSC 3708, Bethesda, MD 20894-3708, USA; Tel: 301-496-0373; Fax: 301-402-6438; E-mail: sidranse@irp.nimh.nih.govCorrespondence: NIH Chemical Genomics Center, NHGRI, NIH, 9800 Medical Center Drive, MSC 3370, Bethesda, MD 20892-3370, USA; Tel: 301-217-5720; Fax: 301-217-5728; E-mail: wzheng@mail.nih.gov
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 3-4-2010 |
Original Manuscript | Fabry Disease – Current Treatment and New Drug Development | |
BACKGROUND
Fabry disease (OMIM 301500) is a hereditary, X-linked lysosomal storage disorder first described by the dermatologists William Anderson and Johann Fabry in 1898 [1Desnick RJ, Ioannou YA, Eng CM. alpha-galactosidase A deficiency: Fabry disease. 8th. New York: McGraw-Hill Professional 2001; 1: p. 1.]. The incidence of the disease is estimated to be 1 in 40,000 to 117,000 males worldwide [1Desnick RJ, Ioannou YA, Eng CM. alpha-galactosidase A deficiency: Fabry disease. 8th. New York: McGraw-Hill Professional 2001; 1: p. 1., 2Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders JAMA 1999; 281: 249-54.]. The disorder is caused by a deficiency of the lysosomal enzyme α-galactosidase A (GLA), which results in the accumulation of the glycosphingolipid, globotriaosylceramide (Gb3), in different cells and organs, notably in endothelial cells and smooth muscle cells of blood vessels [1Desnick RJ, Ioannou YA, Eng CM. alpha-galactosidase A deficiency: Fabry disease. 8th. New York: McGraw-Hill Professional 2001; 1: p. 1.]. The onset of symptoms usually occurs in childhood. By middle age, life-threatening complications often develop in untreated patients. Manifestations of this disease are not only encountered in affected hemizygous males, but are also seen in heterozygous females [3MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females J Med Genet 2001; 38: 769-5., 4Whybra C, Kampmann C, Willers I, et al. Anderson-fabry disease: clinical manifestations of disease in female heterozygotes J Inherit Metab Dis 2001; 24: 715-24.]. Patients with Fabry disease present clinically with chronic neuronopathic pain, gastrointestinal disturbances, a specific skin finding called angiokeratoma, progressive renal impairment, cardiomyopathy, premature myocardial infarctions, and stroke. Both life expectancy and quality of life are severely compromised.
To date, 431 mutations have been reported for the GLA gene (Human Gene Mutation Database, www.hgmd.cf.ac. uk). Of these, more than 57% are missense mutations. Patients with missense mutations often have some residual enzyme activity, ranging from 2% to 25% [1Desnick RJ, Ioannou YA, Eng CM. alpha-galactosidase A deficiency: Fabry disease. 8th. New York: McGraw-Hill Professional 2001; 1: p. 1.]. Studies of the residual GLA activity of mutant forms of the enzyme revealed that many had kinetic properties similar to the wild-type enzyme, but were significantly less stable [5Ishii S, Chang HH, Kawasaki K, et al. Mutant alpha-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin Biochem J 2007; 406: 285-95.-7Ishii S, Kase R, Sakuraba H, Suzuki Y. Characterization of a mutant alpha-galactosidase gene product for the late-onset cardiac form of Fabry disease Biochem Biophys Res Commun 1993; 197: 1585-9.]. These results suggest that the compromised enzyme activity in many Fabry patients is due to protein misfolding, and/or the inability to traffic the enzyme to the lysosomes. Most likely, the mutant protein is retained in the ER and degraded prematurely. Therefore, the discovery of small molecules that assist mutant enzymes to fold correctly may rescue them from premature degradation and increase the amount of active enzyme in the lysosomes.
CURRENT TREATMENTS FOR FABRY DISEASE
Enzyme Replacement Therapy
Enzyme replacement therapy (ERT) supplies recombinant GLA to cells and reverses several of the metabolic and pathologic abnormalities. ERT has been available for the treatment of Fabry disease since 2001 and is administered intravenously once every two weeks [8Brady RO. Enzyme replacement for lysosomal diseases Annu Rev Med 2006; 57: 283-96., 9Lidove O, Joly D, Barbey F, et al. Clinical results of enzyme replacement therapy in Fabry disease: a comprehensive review of literature Int J Clin Pract 2007; 61: 293-302.]. The two recombinant GLA preparations available for ERT are agalsidase alfa (Replagal, Shire Human Genetic Therapies, Cambridge, MA, 0.2 mg/kg per infusion), and agalsidase beta (Fabrazyme, Genzyme Corporation, Cambridge, MA, 1 mg/kg per infusion). However, only Fabrazyme is approved by the FDA for use in the USA [10Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase A--replacement therapy in Fabry's disease N Engl J Med 2001; 345: 9-16.]. ERT has been shown to have a positive effect on kidney and heart manifestations at an early phase of the disease, lessening pain and improving quality of life [9Lidove O, Joly D, Barbey F, et al. Clinical results of enzyme replacement therapy in Fabry disease: a comprehensive review of literature Int J Clin Pract 2007; 61: 293-302.]. However, the long-term clinical benefits of ERT for Fabry patients are still unclear, especially regarding its ability to prevent premature strokes [11Wilcox WR, Banikazemi M, Guffon N, et al. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease Am J Hum Genet 2004; 75: 65-74., 12Moore DF, Scott LT, Gladwin MT, et al. Regional cerebral hyperperfusion and nitric oxide pathway dysregulation in Fabry disease: reversal by enzyme replacement therapy Circulation 2001; 104: 1506-2.]. Some patients also develop immune responses to infused recombinant enzymes [11Wilcox WR, Banikazemi M, Guffon N, et al. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease Am J Hum Genet 2004; 75: 65-74.]. The short half-life of the enzyme and the need for repeated administration of large amounts of enzyme are other limitations of ERT [13Pierre-Louis B, Kumar A, Frishman WH. Fabry disease: cardiac manifestations and therapeutic options Cardiol Rev 2009; 17: 31-5.]. Moreover, the extremely high cost of lifelong treatment becomes a burden to patients with Fabry disease.
Alternative Therapies
While ERT is the standard treatment strategy, many symptoms of Fabry disease can be managed through supportive and palliative approaches. Daily prophylactic doses of neuropathic pain agents, such as phenytoin [14Lockman LA, Hunninghake DB, Krivit W, Desnick RJ. Relief of pain of Fabry's disease by diphenylhydantoin Neurology 1973; 23: 871-5.], carbamazepine [15Lenoir G, Rivron M, Gubler MC, Dufier JL, Tome FS, Guivarch M. Fabry's disease. Carbamazepine therapy in acrodyniform syndrome Arch Fr Pediatr 1977; 34: 704-16.], and gabapentin [16Germain DP. Fabry's disease (alpha-galactosidase-A deficiency): recent therapeutic innovations J Soc Biol 2002; 196: 183-90.] are effective in decreasing the frequency and severity of pain episodes in many patients. Some patients need more potent analgesics, such as opioids, for pain management. For gastrointestinal disturbances, pancrelipase, metoclopramide, H2 blockers, loperamide, and hydrochloride can be beneficial [17Argoff CE, Barton NW, Brady RO, Ziessman HA. Gastrointestinal symptoms and delayed gastric emptying in Fabry's disease: response to metoclopramide Nucl Med Commun 1998; 19: 887-91.]. Various laser treatments for angiokeratomas have been attempted, but have not been helpful [18Mohrenschlager M, Braun-Falco M, Ring J, Abeck D. Fabry disease: recognition and management of cutaneous manifestations Am J Clin Dermatol 2003; 4: 189-96.]. Instead, pedunculated lesions may be treated with liquid nitrogen prior to laser therapy [19Eng CM, Germain DP, Banikazemi M, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement Genet Med 2006; 8: 539-48.].
Therapeutic management primarily focuses on the control of blood pressure, lipids, and proteinuria [20Remuzzi G, Schieppati A, Ruggenenti P. Clinical practice. Nephropathy in patients with type 2 diabetes N Engl J Med 2002; 346: 1145-51., 21Gaede P, Vedel P, Larsen N, Jensen GV, Parving HH, Pedersen O. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes N Engl J Med 2003; 348: 383-93.]. ACE inhibitors and/or blockers should be used in patients with proteinuria [16Germain DP. Fabry's disease (alpha-galactosidase-A deficiency): recent therapeutic innovations J Soc Biol 2002; 196: 183-90.]. Hypertension and hypercholesterolemia should be managed appropriately. Prophylaxis with anticoagulants is important in patients who have had ischemic attacks or stroke [22Desnick RJ, Brady R, Barranger J, et al. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy Ann Intern Med 2003; 138: 338-46.], and permanent cardiac pacing should be considered in high-risk patients [19Eng CM, Germain DP, Banikazemi M, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement Genet Med 2006; 8: 539-48.]. Furthermore, patients need to be encouraged to maintain a healthy lifestyle. A change in eating habits, which includes frequent small meals, can be helpful [16Germain DP. Fabry's disease (alpha-galactosidase-A deficiency): recent therapeutic innovations J Soc Biol 2002; 196: 183-90.]. While renal failure is the most frequent cause of death in classic Fabry disease, in patients with advanced renal disease, dialysis or transplantation can prolong life [23Donati D, Sabbadini MG, Capsoni F, et al. Immune function and renal transplantation in Fabry's disease Proc Eur Dial Transplant Assoc Eur Ren Assoc 1985; 21: 686-92.]. However, even with the engrafted kidneys, other organ system damage continues, particularly vascular disease affecting the heart and brain [24Kramer W, Thormann J, Mueller K, Frenzel H. Progressive cardiac involvement by Fabry's disease despite successful renal allotransplantation Int J Cardiol 1985; 7: 72-5.]. It is clear that even with ERT, other treatments and preventative measures are necessary to manage Fabry disease.
EMERGING TREATMENT STRATEGIES FOR FABRY DISEASE
Emerging treatment strategies for Fabry disease involve the development of small molecule compounds, which are widely used for the treatment of a variety of diseases. Such small molecules account for approximately 80-90% of marketed drugs. Small molecule drugs are usually effective, have quick responses, and can be administered orally. In addition, they can cross the blood-brain barrier, do not cause autoimmune responses, and have lower manufacturing costs. Gene therapy has also been explored for the treatment of Fabry disease. The experimental results were limited as the viral vectors were used to achieve in vivo gene transfer in murine models [25Sands MS, Davidson BL. Gene therapy for lysosomal storage diseases Mol Ther 2006; 13: 839-49.]. Some emerging drug development strategies for small molecule therapy of Fabry disease are illustrated in Fig. (1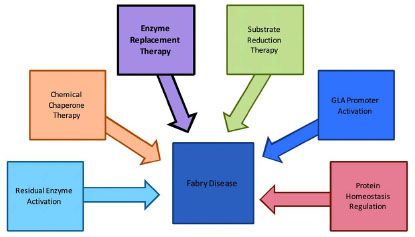 ) and outlined in more detail below.
) and outlined in more detail below.
 |
Fig. (1) Illustration of current and emerging treatment strategies for Fabry disease. The currently available treatment is highlighted in black. |
Substrate Reduction Therapy
GLA hydrolyzes the terminal alpha-galactosyl moieties from glycosphingolipids. A deficiency in GLA causes the accumulation of the glycosphingolipid, globotriaosylceramide, in lysosomes. Substrate reduction therapy (SRT) circumvents enzyme replacement/modification by inhibiting synthesis of globotriaosylceramide. This approach involves the use of a glucosylceramide synthase inhibitor, which would slow the rate of Gb3 synthesis, and thus decrease lysosomal storage. Even if GLA activity is low or undetectable, SRT may convert a severe disease phenotype to a milder one. N-butyldeoxynojirimycin (NB-DNJ), an iminosugar analog, has been used as a glucosylceramide synthase inhibitor [26Dwek RA, Butters TD, Platt FM, Zitzmann N. Targeting glycosylation as a therapeutic approach Nat Rev Drug Discov 2002; 1: 65-75.]. However, the inhibitory effect of NB-DNJ is not very specific for glucosylceramide synthase and side effects are observed, including gastrointestinal complaints [27Cox T, Lachmann R, Hollak C, et al. Novel oral treatment of Gaucher's disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis Lancet 2000; 355: 1481-5., 28Elstein D, Hollak C, Aerts JM, et al. Sustained therapeutic effects of oral miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher disease J Inherit Metab Dis 2004; 27: 757-66.]. Since ERT for Fabry disease only shows modest efficacy, combinatorial therapy using ERT and SRT is being considered as a treatment strategy.
Residual Enzyme Activation
It is known that enzyme activity can be stimulated by small molecule activators. Enzyme activators may increase the residual activity of mutant GLA in the lysosomes of patients with Fabry disease, thereby lessening lysosomal storage of the substrate and alleviating symptoms. However, these activators may not be beneficial if their efficacy is not high enough or if the residual activity of mutant GLA is inadequate. From the drug discovery point of view, it is much more difficult to identify enzyme activators than inhibitors in compound library screens. Therefore, assays need to be developed specifically for the identification of small molecule enzyme activators.
GLA Promoter Activation
Specific small molecule promoter activators may increase the amount of GLA in lysosomes by stimulating expression of the target protein. Here, a promoter activator binds the GLA promoter in the nuclei of cells and enhances GLA transcription, thereby increasing the synthesis of mutant GLA protein. This results in an increased amount of GLA in the lysosomes, as the enhancement of mutant enzyme expression could proportionally increase protein trafficking to the lysosome [29Gelsthorpe ME, Baumann N, Millard E, et al. Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding J Biol Chem 2008; 283: 8229-36.]. Therefore, in Fabry patients with significant residual GLA enzyme activity, a small molecule promoter activator may correct lysosomal storage by amplifying the amount of enzyme in lysosomes. In order to identify small molecule GLA promoter activators, a permanent cell line transformed with a cDNA plasmid containing the promoter region of the GLA gene linked with a reporter gene such as luciferase has to be established. Promoter activator lead compounds may be found by high throughput screening of this reporter gene cell line against the compound library. Once lead compounds are identified, chemistry optimization and compound development can begin.
Protein Homeostasis Regulation (Proteostasis)
Another treatment strategy involves altering the proteostasis network in cells, which consists of many highly regulated biological pathways that influence protein synthesis, folding, trafficking, disaggregation and degradation [30Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention Science 2008; 319: 916-.]. Small molecule proteostasis regulators may be utilized to increase the amount of folded protein in the endoplasmic reticulum by increasing the proteostasis network capacity. This is achieved through the enhancement of signaling pathways and/or the transcription and translation of components in the proteostasis network [31Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency Annu Rev Biochem 2009; 78: 959-1.]. Such increases and decreases in the proteostasis network capacity can promote either protein folding or degradation, respectively. It has recently been reported that diltiazem and verapamil, known calcium channel blockers, partially restored mutant glucocerebrosidase folding, trafficking, and enzyme function in patient fibroblasts [32Mu TW, Fowler DM, Kelly JW. Partial restoration of mutant enzyme homeostasis in three distinct lysosomal storage disease cell lines by altering calcium homeostasis PLoS Biol 2008; 6: e26.]. Although therapeutic concentrations of these calcium channel blockers cannot be reached in humans, there is potential for small molecule development using this new target. In addition, the combination of proteostasis regulators with small molecule chaperones may further increase the amount of folded protein trafficked to lysosomes and thus enhance the therapy, although this hypothesis needs to be tested.
Chemical Chaperone Therapy
Chemical chaperone therapy (CCT) has recently emerged as a potential therapeutic alternative for Fabry disease. Chemical chaperones are small molecules that bind to mutant enzyme proteins and assist in their correct folding, maturation, and trafficking to their functional site, such as the lysosome. It has been reported that some enzyme inhibitors and receptor antagonists can serve as chaperones for mutant enzymes and receptors, as they bind to the target protein tightly [33Bernier V, Lagace M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases Trends Endocrinol Metab 2004; 15: 222-8., 34Ulloa-Aguirre A, Janovick JA, Brothers SP, Conn PM. Pharmacologic rescue of conformationally-defective proteins: implications for the treatment of human disease Traffic 2004; 5: 821-37.]. The effects of chemical chaperones have been explored for various lysosomal storage disorders, including Gaucher disease [35Sawkar AR, Cheng WC, Beutler E, Wong CH, Balch WE, Kelly JW. Chemical chaperones increase the cellular activity of N370S beta -glucosidase: a therapeutic strategy for Gaucher disease Proc Natl Acad Sci USA 2002; 99: 15428-33.-38Zheng W, Padia J, Urban DJ, et al. Three classes of glucocerebrosidase inhibitors identified by quantitative high-throughput screening are chaperone leads for Gaucher disease Proc Natl Acad Sci USA 2007; 104: 13192-7.], Pompe disease [39Okumiya T, Kroos MA, Vliet LV, Takeuchi H, Van der Ploeg AT, Reuser AJ. Chemical chaperones improve transport and enhance stability of mutant alpha-glucosidases in glycogen storage disease type II Mol Genet Metab 2007; 90: 49-57., 40Parenti G, Zuppaldi A, Gabriela Pittis M, et al. Pharmacological enhancement of mutated alpha-glucosidase activity in fibroblasts from patients with Pompe disease Mol Ther 2007; 15: 508-14.], Tay-Sachs/Sandhoff disease [41Tropak MB, Reid SP, Guiral M, Withers SG, Mahuran D. Pharmacological enhancement of beta-hexosaminidase activity in fibroblasts from adult Tay-Sachs and Sandhoff Patients J Biol Chem 2004; 279: 13478-87.], GM1-gangliosidosis [42Matsuda J, Suzuki O, Oshima A, et al. Chemical chaperone therapy for brain pathology in G(M1)-gangliosidosis Proc Natl Acad Sci USA 2003; 100: 15912-7.], as well as for Fabry disease. Recent studies have shown that chemical chaperones can improve the efficacy of enzyme replacement therapy for Gaucher disease, Pompe disease, and Fabry disease [43Shen JS, Edwards NJ, Hong YB, Murray GJ. Isofagomine increases lysosomal delivery of exogenous glucocerebrosidase Biochem Biophys Res Commun 2008; 369: 1071-5., 44Porto C, Cardone M, Fontana F, et al. The pharmacological chaperone N-butyldeoxynojirimycin enhances enzyme replacement therapy in Pompe disease fibroblasts Mol Ther 2009; 17: 964-71.]. One GLA inhibitor, 1-deoxygalactonojirimycin (DGJ, marketed as AmigalTM by Amicus Therapeutics, Inc.) is currently being studied in a phase 3 clinical trial as a chaperone therapeutic agent for Fabry disease [45Fan JQ, Ishii S. Active-site-specific chaperone therapy for Fabry disease. Yin and Yang of enzyme inhibitors FEBS J 2007; 274: 4962-71.].
However, increasing mutant enzyme activities with an inhibitor may not be an ideal solution in vivo, as the chaperone action may be functionally compromised by the inhibition of enzyme activity. In theory, an enzyme activator or a pure enzyme binder with chaperone activity would be a better choice as a candidate for drug development. If an enzyme activator is used as a therapy, both the chaperone and enzyme stimulatory actions of the activator would synergistically increase enzyme activity in the lysosomes. Currently, no such small molecule activators with chaperone activity are available. The proper assay development and compound library screens for identifying new lead compounds remain to be explored. Cell based chaperone assays using tagged GLA proteins may be a useful new method to identify potential chaperone compounds in library screens.
THE PROCESS OF SMALL MOLECULE DRUG DISCOVERY
Overview
The drug discovery process has evolved from disease model based screens with low throughput, to mechanism/molecular target based screens with high throughput in the last two decades. The current small molecule drug discovery process includes target identification and validation, early lead discovery and optimization, preclinical safety development, and clinical trials, and requires an average of 10 to 15 years (Fig. 2 ). Modern target based drug discovery started in the late 1980s, when advancements in molecular biology and the Human Genome Project led to a wealth of new drug targets. Recombinant proteins/enzymes and target proteins expressed in cell lines became available as useful tools for identifying small molecule activators and inhibitors in compound library screens. The demand for the high throughput screening of molecular targets led to the rapid development of new assay/detection technologies and drug screening robots in the 1990s. Synthetic chemical compounds for compound collections also became commercially available at reasonable costs. A screen using 1-3 million compounds to identify lead compounds for a single drug target has become a routine operation in the pharmaceutical industry over the last 10 years.
). Modern target based drug discovery started in the late 1980s, when advancements in molecular biology and the Human Genome Project led to a wealth of new drug targets. Recombinant proteins/enzymes and target proteins expressed in cell lines became available as useful tools for identifying small molecule activators and inhibitors in compound library screens. The demand for the high throughput screening of molecular targets led to the rapid development of new assay/detection technologies and drug screening robots in the 1990s. Synthetic chemical compounds for compound collections also became commercially available at reasonable costs. A screen using 1-3 million compounds to identify lead compounds for a single drug target has become a routine operation in the pharmaceutical industry over the last 10 years.
 |
Fig. (2) Schematic of the drug discovery/development process. |
Compound screening methods and throughput have significantly improved in the last 20 years. The initial test tube (1-5 ml) based screens have evolved to 96-well plates (100-300 µl), and recently to high density 384-well (20-40 µl) and 1536-well (2-8 µl) plates. The increase in well density has greatly enhanced compound screening throughput, and has also reduced screening costs. Primary screens traditionally use a single compound concentration due to the large size of libraries (1-3 million compounds). Recently, a quantitative high throughput screening (qHTS) method has been developed , where compounds are screened at a 1:5 titration, and concentration-response curves are obtained for active compounds immediately after the primary screen [46Inglese J, Auld DS, Jadhav A, et al. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries Proc Natl Acad Sci USA 2006; 103: 11473-78.]. A primary screen with multiple compound concentrations increases the data quality, reduces the number of compounds being selected for confirmation, and is particularly helpful for identifying biphasic compounds that might be missed in a single concentration screen. However, the qHTS method may not be applicable in all screen scenarios as increasing reagent costs may prevent multiple concentration screens. Reduction in the library size to 200,000- 400,000 compounds, use of an alternative assay, or further assay miniaturization could bring down reagent costs and make multiple concentration screens more practical.
Once active compounds (“hits”) are identified from the primary screen, they must be tested further in secondary and tertiary assays to confirm their activity and selectivity. The compounds with the most drug-like chemical structures then undergo chemical optimization to improve potency, selectivity and reduce potential toxicity. Hundreds to thousands of newly synthesized analog compounds are rescreened to determine structure-activity-relationships (SAR) as well as pharmacokinetic properties, such as drug absorption, distribution, metabolism, and excretion properties. The compounds with the highest potencies, selectivities, and most promising pharmacokinetic properties then move on to preclinical development, where safety and efficacy in animal models is tested, before entering clinical trials. It is clear that target selection, assay development, and lead discovery play key roles in the drug discovery and development process. Identification of high quality, drug-like lead compounds is vital for success in the later stages of drug development.
Assay Development
Primary screening assays should have the least possible assay steps (usually 3-6 steps), have a short incubation time (from a few minutes to a maximum of 3 days), be homogenous (no wash step), have high repeatability, have physiological relevance, and reagents should have a reasonable cost. For lysosomal hydrolases, including GLA, fluorescence assays are commonly used. Fluorogenic substrates, such as 4-methylumbelliferyl-α-D-galactopyranoside (4MU-α-Gala) [47Mapes CA, Sweeley CC. Galactosyl (alpha 1--4)galactosylceramide: galactosyl hydrolase activity in normal and Fabry plasma Biochem Biophys Res Commun 1973; 53: 1317-24., 48Hultberg B, Sjoblad S, Ockerman PA. Glycosidases in human skin fibroblast cultures. Alpha-fucosidase, alpha-galactosidase, alphaglucosidase, beta-mannosidase, and N-acetyl-alphaglucosaminidase. Acta Paediatr Scand 1975; 64: 123-31.] or resorufinyl α-D-galactopyranoside (res-α-Gala) [49Shi ZD, Motabar O, Goldin E, et al. Synthesis and characterization of a new fluorogenic substrate for alpha-galactosidase Anal Bioanal Chem 2009; 394(7): 1903-9.], can be used to measure GLA activity. Chromogenic substrates, such as p-nitrophenol-α-D-galactopyranoside [50de Groot PG, Hamers MN, Westerveld A, et al. A new immunochemical method for the quantitative measurement of specific gene products in man-rodent somatic cell hybrids Hum Genet 1978; 44: 295-304., 51Yagi F, Eckhardt AE, Goldstein IJ. Glycosidases of Ehrlich ascites tumor cells and ascitic fluid--purification and substrate specificity of alpha-N-acetylgalactosaminidase and alpha-galactosidase: comparison with coffee bean alpha-galactosidase Arch Biochem Biophys 1990; 280: 61-7.] or naphthyl-α-D-galactopyranoside [52Tsou KC, Su HC. A study of yeast alpha-galactosidase with naphthyl alpha-D-galactopyranosides as chromogenic substrates Anal Biochem 1964; 8: 415-23.], are also available. However, absorbance assays are usually less sensitive than fluorescence assays, and cannot be easily miniaturized into a 1536-well plate format.
During assay development, various conditions, such as the buffer components, pH, incubation time and temperature, enzyme and substrate concentrations, and DMSO tolerance have to be evaluated and optimized prior to HTS. Since GLA is a lysosomal enzyme, an acidic buffer should be used. The incubation time should give sufficient fluorescence signal while being in the linear portion of the time course curve. To increase assay sensitivity, the minimum amount of enzyme which produces sufficient signal should be used, and the substrate concentration should be around or below the Km value. The effect of DMSO on enzyme activity is also important because it is commonly used as a solvent to dissolve compounds. Usually the concentration of DMSO in the assay should be less than 1%.
Assay Validation
Known inhibitors or activators are commonly used to validate assay sensitivity, and to compare the assay in different plate formats, such as in 96-, 384- and 1536-well plate formats. Potencies (IC50 or EC50 values) of the control compounds should usually be within two fold of the reported value. A DMSO plate (no compounds) is used to determine the signal-to-basal (S/B) ratio, CV, and the Z’ factor of the assay. The S/B ratio (assay window) should be greater than 2, and the CV should be smaller than 10%, but no greater than 20%. The Z’ factor is a parameter used to judge the quality of a screening assay, with a value greater than 0.5 being acceptable for HTS. It is calculated using the following equation: Z’ = 1 – (3*s.d.(1) + 3*s.d.(2))/(A-B), where s.d.(1) is the standard deviation of enzyme activity, s.d.(2) is the standard deviation of basal activity (without enzyme), A is the mean of enzyme activity and B is the mean of basal activity. Reagent stability over a 24 hour time period (enzyme and substrate) must be examined to ensure that the signal-to-basal ratio remains nearly constant throughout the continuous robotic screen. Usually, an enzyme assay with an S/B ratio greater than 3, CV less than 10% and Z’ factor greater than 0.5 is considered a robust screening assay. An S/B ratio of at least 2 is usually necessary for HTS.
After the DMSO plate test, a test screen with a smaller compound library is carried out to validate the screen performance of the optimized assay. The LOPAC collection (Library of Pharmacologically Active Compounds, Sigma-Aldrich), which consists of 1,280 compounds, is commonly used for this screening test [46Inglese J, Auld DS, Jadhav A, et al. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries Proc Natl Acad Sci USA 2006; 103: 11473-78.]. The S/B ratio, Z’ and the hit rate of the primary screen can be assessed by this test screen. Since the LOPAC collection contains compounds that interact with a broad range of targets, the hit rate obtained is usually higher than that obtained from the high throughput screen, which uses a much larger compound collection. However, this test screen is helpful in predicting the outcome of the large-scale compound screen.
Large-Scale Compound Screen (HTS)
Once the assay is validated using a DMSO plate and the LOPAC library, and the results indicate that the assay is of sufficient quality (S/B ratio greater than 2 and Z’ factor greater than 0.5), a full library screen using a robot is ready to be performed. HTS consumes a significant quantity of reagents, as several hundred to a few thousand plates are screened, depending on the size of the compound libraries and the assay conditions. The robotic screen is usually run continuously, which requires the reagents to be stable at room temperature or 4°C for at least 24 hours. Depending on the size of the compound collection, the screening can take anywhere from a few days to two weeks.
After the primary screen is complete, the data is loaded into a database for analysis. Traditionally, the percent inhibition or activation is used to calculate the hit rate at the compound concentration selected for the screen. The expected hit rate is usually 0.1-1% of the compound collection. However, selection of active compounds from qHTS is based on the potencies (EC50/IC50) and the quality of the concentration response curves. In addition, structure clustering and early SAR analysis can be performed immediately on the primary screen results, which reduces the selection or “cherry-picking” of too many compounds for hit confirmation.
Confirmation and Secondary Screens
The hits from the primary screen are confirmed by testing them in the same assay. Once confirmed as hits, the compounds are then tested for specificity. In the case of GLA, other lysosomal hydrolases can be used to assess compound selectivity, such as glucocerebrosidase, α-glucosidase, and β-N-acetylglucosaminidase. Like the GLA assay, these counterscreen assays are fluorogenic and can be performed in a similar assay format. Because the same fluorophore is used in these counterscreen assays, false positive compounds, including fluorescence quenchers and fluorescent compounds, can be eliminated. The ideal lead compound should have at least ten times less activity in the counterscreen assays.
The mode of inhibition of the lead inhibitors should be characterized in an enzyme kinetic assay. A series of 5-6 substrate concentration response curves in the absence and presence of several concentrations of inhibitors are generated kinetically. A Lineweaver–Burk plot (double reciprocal plot) is commonly used to determine if the inhibition is competitive, noncompetitive, or uncompetitive. Ki values can be obtained by using a computer program such as Prism® (Graphpad, San Diego, CA). A rapid Ki assessment method has been developed, permitting measurement of the Ki values of 6 compounds in one 1536-well plate using a fluorogenic enzyme assay [38Zheng W, Padia J, Urban DJ, et al. Three classes of glucocerebrosidase inhibitors identified by quantitative high-throughput screening are chaperone leads for Gaucher disease Proc Natl Acad Sci USA 2007; 104: 13192-7.].
Additional assays are needed to further characterize the lead compounds. In the case of GLA, the compound’s potential to be a chemical chaperone should be evaluated. This usually can be done either by determining if the lead compounds are able to increase enzyme activity in whole cells or by visualizing decreased substrate storage or increased GLA protein in lysosomes.
Whole Cell Enzyme Activity Assay
A whole cell enzyme activity assay can be used to test for potential GLA chaperoning. This involves the use of patient derived primary skin fibroblasts or lymphoblasts to measure the effect of the compounds on enzymatic activity. Briefly, the cells are cultured with or without the compounds in standard cell culture conditions for 2-5 days, and then GLA activity is measured in whole cells using a fluorogenic substrate, either 4MU-α -Gala or res-α -Gala. The substrates enter the cells via permeablization of cell membranes by 0.2 M sodium acetate-PBS buffer (1:1 ratio) at pH 4.0 [38Zheng W, Padia J, Urban DJ, et al. Three classes of glucocerebrosidase inhibitors identified by quantitative high-throughput screening are chaperone leads for Gaucher disease Proc Natl Acad Sci USA 2007; 104: 13192-7.]. Compounds capable of enhancing GLA activity in patient cells would be considered potential chemical chaperones.
Immunofluorescence Staining Assay
This assay involves the double immunofluorescence staining of Gb3 and the lysosomal membrane protein 1 (LAMP1) after 2-5 days compound treatment [53Yam GH, Bosshard N, Zuber C, Steinmann B, Roth J. Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants Am J Physiol Cell Physiol 2006; 290: C1076-82.]. Compounds with chaperone activity would reduce Gb3 storage in the lysosome, and thus, Gb3 staining in cells should become distinctively diminished after treatment with active compounds. Also, an anti-GLA antibody can be used to determine whether the compounds acted as chaperones, as reflected by the degree of co-localization with LAMP1 [54Yam GH, Zuber C, Roth J. A synthetic chaperone corrects the trafficking defect and disease phenotype in a protein misfolding disorder FASEB J 2005; 19: 12-8.]. Lysotracker (Invitrogen), a fluorescent dye staining to the acidic compartments in cells, can be used to visualize the enlarged lysosomes. Chaperone compounds would increase the amount of GLA in lysosomes, thereby increasing cleavage of the substrate. As a result, smaller lysosomes would be observed though the false positive compounds which alter the intracellular pH should be eliminated.
Natural Substrate Assay
The natural substrate for GLA, globotriaosylceramide (Gb3), is used to assess the compounds in a more physiologically relevant environment. Several methods have been reported to measure Gb3, including gas chromatography/chemical ionization mass spectrometry (GC/CI-MS) after permethylation of glycolipids [55Levery SB, Hakomori S. Microscale methylation analysis of glycolipids using capillary gas chromatography-chemical ionization mass fragmentography with selected ion monitoring Methods Enzymol 1987; 138: 13-25.], HPLC with UV detection after perbenzoylation [56Oshima M, Asano K, Shibata S, Suzuki Y, Masuzawa M. Urinary neutral glycosphingolipid analysis of patients with Fabry's disease; rapid isocratic elution from high-performance liquid chromatography as per-o-benzoyl derivatives Biochim Biophys Acta 1990; 1043: 157-60.], and enzyme-linked immunosorbent assay [57Zeidner KM, Desnick RJ, Ioannou YA. Quantitative determination of globotriaosylceramide by immunodetection of glycolipid-bound recombinant verotoxin B subunit Anal Biochem 1999; 267: 104-3.]. Recently, a more sensitive and robust method has been developed which uses electrospray ionization mass spectrometry (ESI-MS) [58Fauler G, Rechberger GN, Devrnja D, et al. Rapid determination of urinary globotriaosylceramide isoform profiles by electrospray ionization mass spectrometry using stearoyl-d35-globotriaosylceramide as internal standard Rapid Commun Mass Spectrom 2005; 19: 1499-506.]. However, this method still has a relatively low throughput, and would only be useful for confirming the activity of a small number of compounds.
GLA Western Blot
Western Blot analyses of patient-derived cells can be compared with normal cells to evaluate GLA protein maturation after treatment with compounds. GLA is present in two main forms within cells, the immature ER form (51 kDa) and the mature lysosomal form (46 kDa) [53Yam GH, Bosshard N, Zuber C, Steinmann B, Roth J. Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants Am J Physiol Cell Physiol 2006; 290: C1076-82., 59Fan JQ, Ishii S, Asano N, Suzuki Y. Accelerated transport and maturation of lysosomal alpha-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor Nat Med 1999; 5: 112-5., 60Shin SH, Murray GJ, Kluepfel-Stahl S, et al. Screening for pharmacological chaperones in Fabry disease Biochem Biophys Res Commun 2007; 359: 168-73.]. The intensity of each band on the Western Blot, and the relative ratio of the two bands, provides information about protein expression and processing.
Probe Identification and Optimization
Depending on the potency and selectivity of active compounds identified in the above processes, a probe can be defined if the potencies (IC50/EC50) of the identified agents are less than 1 µM, and if they selectively act on GLA and effectively reduce Gb3 accumulation in patient cells. If the activity of the compounds is weak and/or the specificity poor, probe optimization can be carried out by chemists. The bioavailability of the active compound(s) is also optimized during this process.
Further Testing in Animal Models
Several mouse models have been reported for Fabry disease. A complete GLA knockout model was generated that shows some Gb3 accumulation in various organs and tissues [61Ohshima T, Murray GJ, Swaim WD, et al. alpha-Galactosidase A deficient mice: a model of Fabry disease Proc Natl Acad Sci USA 1997; 94: 2540-4.]. This model has been used for experiments with ERT, SRT, and gene therapy. However, this model is not useful for examining the effect of chemical chaperones because it has no mutant enzyme that can be enhanced by chaperones. However, a GLA knock-in mouse that contains the human mutation R301Q has been generated that may prove useful in answering this question [62Ishii S, Yoshioka H, Mannen K, Kulkarni AB, Fan JQ. Transgenic mouse expressing human mutant alpha-galactosidase A in an endogenous enzyme deficient background: a biochemical animal model for studying active-site specific chaperone therapy for Fabry disease Biochim Biophys Acta 2004; 1690: 250-7.]. Because the expression level of the human transgene is substantially higher than that of the endogenous gene, this mouse has no signs or symptoms of Fabry disease. However, it has been shown that the activity of the mutant GLA can be enhanced by DGJ in vivo [62Ishii S, Yoshioka H, Mannen K, Kulkarni AB, Fan JQ. Transgenic mouse expressing human mutant alpha-galactosidase A in an endogenous enzyme deficient background: a biochemical animal model for studying active-site specific chaperone therapy for Fabry disease Biochim Biophys Acta 2004; 1690: 250-7.]. Mouse models with mutations that still retain some enzyme activity will prove useful for testing probes identified from HTS.
CONCLUSIONS
In summary, this review provides an overview of the current and emerging therapies for Fabry disease, and describes drug development strategies and methods. Although enzyme replacement therapy is effective, there is a need for other therapeutic strategies, which can either serve as primary or supplemental treatments. Small molecule drug discovery is promising, as it could lead to new treatments for Fabry disease. The discovery of non-inhibitory chaperones, activators, or inhibitors of the enzymes that degrade glycosphingolipids, would be a major breakthrough. The continued expansion of our knowledge regarding the biology and pathophysiology of Fabry disease, combined with rapid advances in drug discovery technologies, bring us closer to the discovery of new treatments for Fabry disease and give hope to people suffering from this complex and life-threatening disease.
ACKNOWLEDGEMENTS
This work was supported by the Intramural Research Programs of the National Human Genome Research Institute and National Institutes of Health and by the Molecular Libraries Initiative of the NIH Roadmap for Medical Research.