- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Open Chemistry Journal
(Discontinued)
ISSN: 1874-8422 ― Volume 8, 2021
A Variable Temperature Study of the π–π Stacking Interaction in the Co-Crystal Naphthalene-Octafluoronaphthalene
Judith Burrows1, Prithwish Sain1, Graham C. Saunders1, *
Abstract
Introduction:
The structure of the 1:1 co-crystal of naphthalene and octafluoronaphthalene, which has been previously determined at room temperature, was determined at 100, 150, 200 and 250 K.
Results:
Reductions in intermolecular distances and unit cell dimensions are observed on cooling. DFT calculations reveal that the energies of interaction between pairs of molecules vary little with temperature.
Conclusion:
The strongest interaction is the π–π stacking between virtually parallel naphthalene and octafluoronaphthalene molecules and this displays less change with temperature than the other, weaker, interactions, which have much shallower energy minima.
Article Information

Identifiers and Pagination:
Year: 2019Volume: 6
First Page: 66
Last Page: 73
Publisher Id: CHEM-6-66
DOI: 10.2174/1874842201906010066
Article History:
Received Date: 03/10/2019Revision Received Date: 21/10/2019
Acceptance Date: 31/10/2019
Electronic publication date: 31/12/2019
Collection year: 2019
open-access license: This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International Public License (CC-BY 4.0), a copy of which is available at: (https://creativecommons.org/licenses/by/4.0/legalcode). This license permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
* Address correspondence to this author at School of Science, University of Waikato, Private Bag 3105, Hamilton 3240, New Zealand;
E-mail: graham.saunders@waikato.ac.nz
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 03-10-2019 |
Original Manuscript | A Variable Temperature Study of the π–π Stacking Interaction in the Co-Crystal Naphthalene-Octafluoronaphthalene | |
1. INTRODUCTION
The π–π stacking interaction between arenes and polyfluoroarenes is being increasingly use in crystal engineering [1Hori, A. The Importance of Pi-Interactions in Crystal Engineering: Frontiers in Crystal Engineering2012.-13Oosterwijk, M.A.; Saunders, G.C.; Zou, W. Polyfluoroarene-arene π–π stacking in three directions to provide crystal polarity. J. Fluor. Chem., 2016, 188, 80-84.
[http://dx.doi.org/10.1016/j.jfluchem.2016.06.015] ]. A number of studies have indicated its geometric properties, in particular a separation of ca. 3.35 Å between the planes of the virtually parallel arene and polyfluoroarene, and an offset of ca. 1.7 Å [1Hori, A. The Importance of Pi-Interactions in Crystal Engineering: Frontiers in Crystal Engineering2012.-29Williams, J.H.; Cockcroft, J.K.; Fitch, A.N. Structure of the lowest temperature phase of the solid benzene–hexafluorobenzene adduct. Angew. Chem. Int. Ed. Engl., 1992, 31, 1655-1657.
[http://dx.doi.org/10.1002/anie.199216551] ], and strength, which has been calculated as 36 and 47 kJ mol-1 for that between toluene and hexafluorobenzene [30Gung, B.W.; Amicangelo, J.C. Substituent effects in C6F6-C6H5X stacking interactions. J. Org. Chem., 2006, 71(25), 9261-9270.
[http://dx.doi.org/10.1021/jo061235h] [PMID: 17137351] ], and that between naphthalene and octafluoronaphthalene [31Bacchi, S.; Benaglia, M.; Cozzi, F.; Demartin, F.; Filippini, G.; Gavezzotti, A. X-ray diffraction and theoretical studies for the quantitative assessment of intermolecular arene-perfluoroarene stacking interactions. Chemistry, 2006, 12(13), 3538-3546.
[http://dx.doi.org/10.1002/chem.200501248] [PMID: 16506260] ] respectively. The majority of crystal structures of arene-polyfluoroarene co-crystals and molecules which display π–π stacking have been determined either at room temperature [14Naae, D.G. Biphenyl-perfluorobiphenyl; 1:1 molecular complex. Acta Crystallogr., 1979, B35, 2765-2768.
[http://dx.doi.org/10.1107/S0567740879010499] -18Adamson, A.J.; Archambeau, Y.; Banks, R.E.; Beagley, B.; Helliwell, M.; Pritchard, R.G.; Tipping, A.E. p-Methyl-N-(pentafluorobenzylidene)aniline (1), 1,2,3,4-tetrafluoro-7-methoxyacridine (2), 1,2,3,4,7-pentafluoroacridine (3) and 3-(p-methylanilino)-1,2,4-trifluoro-7-methylacridine (4): four molecules representing key stages in the one-pot synthesis of 1,2,3,4-tetrafluoroacridines by treating pentafluorobenzaldehyde with para-substituted anilines. Acta Crystallogr., 1994, 50, 967-971., 32Potenza, J.; Mastropaolo, D. Naphthalene-octafluoronaphthalene, 1:1 Solid Compound. Acta Crystallogr., 1975, B31, 2527-2529.
[http://dx.doi.org/10.1107/S0567740875008011] ] or low temperature [19Day, M.W.; Matzger, A.J.; Grubbs, R.H. 1,3,5-tris(2-Phenylethynyl)benzene tris(hexafluorobenzene)., 2001, -23Grzegorczyk, M.; Gdaniec, M. 2,2′,3,3′,4,4′,5,5′,6,6′-Decafluorodiphenylamine and its 1:1 cocrystal with diphenylamine. Acta Crystallogr. 2006, C62, o419-o422. (b) Stoll, I.; Brodbeck, R.; Neumann, B.; Stammler, H.-G.; Mattay, J. Controlling the self assembly of arene functionalised 2-aminopyrimidines by arene-perfluoroarene interaction and by silver(I) complex formation. CrystEngComm, 2009, 11, 306-317.], but rarely both [24Dahl, T. Crystal structure of the trigonal form of the 1:1 complex between hexamethylbenzene and hexafluorobenzene. Acta Chem. Scand., 1972, 26, 1569-1575.
[http://dx.doi.org/10.3891/acta.chem.scand.26-1569] -29Williams, J.H.; Cockcroft, J.K.; Fitch, A.N. Structure of the lowest temperature phase of the solid benzene–hexafluorobenzene adduct. Angew. Chem. Int. Ed. Engl., 1992, 31, 1655-1657.
[http://dx.doi.org/10.1002/anie.199216551] ], and to our knowledge only one variable temperature structural study has been undertaken, that for the crystal structure of 1,2,3,4-tetrafluoronaphthalene (CCDC reference CAXNUL) [33Bagryanskaya, I.Y.; Gatilov, Y.V.; Maksimov, A.M.; Platonov, V.E.; Zibarev, A.V. Supramolecular synthons in crystals of partially fluorinated fused aromatics: 1,2,3,4-Tetrafluoronaphthalene and its aza-analogue 1,3,4-trifluoroisoquinoline. J. Fluor. Chem., 2005, 126, 1281-1287.
[http://dx.doi.org/10.1016/j.jfluchem.2005.06.011] , 34Cozzi, F.; Bacchi, S.; Filippini, G.; Pilati, T.; Gavezzotti, A. Synthesis, X-ray diffraction and computational study of the crystal packing of polycyclic hydrocarbons featuring aromatic and perfluoroaromatic rings condensed in the same molecule: 1,2,3,4-tetrafluoronaphthalene, -anthracene and -phenanthrene. Chemistry, 2007, 13(25), 7177-7184.
[http://dx.doi.org/10.1002/chem.200700267] [PMID: 17568459] ]. However, although theoretical lattice energies were calculated, changes in the π–π stacking geometry and the energy of interaction were not reported.
In order to address this deficiency, we decided to perform a variable temperature structural study on a suitable co-crystal containing infinite columns of π–π stacked alternating arene and polyfluoroarene molecules. For the study, it would be desirable if the columns, and consequently the π–π stacking interaction, occurs along one crystallographic axis, so that any changes in this direction can be related directly to the interaction. For simplicity it would also be desirable if the interactions were symmetric about each molecule, i.e. the midpoint of each molecule lies on a crystallographic centre of inversion. The crystal structure of naphthalene-octafluoronaphthalene, 1, which has been determined at room temperature [32Potenza, J.; Mastropaolo, D. Naphthalene-octafluoronaphthalene, 1:1 Solid Compound. Acta Crystallogr., 1975, B31, 2527-2529.
[http://dx.doi.org/10.1107/S0567740875008011] ] (CCDC reference NPOFNP) satisfies both criteria. Here, we report the results of the study.
2. RESULTS AND DISCUSSION
The crystal structure of naphthalene-octafluoronaphthalene co-crystal, 1, was determined at 100, 150, 200 and 250 K, all of which are consistent with that previously reported for room temperature [32Potenza, J.; Mastropaolo, D. Naphthalene-octafluoronaphthalene, 1:1 Solid Compound. Acta Crystallogr., 1975, B31, 2527-2529.
[http://dx.doi.org/10.1107/S0567740875008011] ] (CCDC reference NPOFNP). Co-crystal 1 crystallizes in the P21/c space group, with columns of alternating parallel naphthalene and octafluoronaphthalene molecules, parallel to the a axis (Fig. 1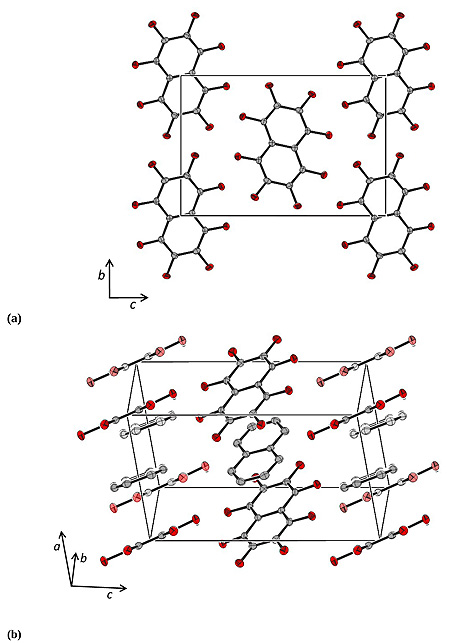 ), with the midpoints of all the molecules lying on centres of inversion. The crystal data are presented in Table 1, selected bond distances and angles in Table 2, and selected intermolecular distances and angles in Table 3. The molecular structure of 1 at 100 K and the atom numbering scheme are shown in Fig. (2
), with the midpoints of all the molecules lying on centres of inversion. The crystal data are presented in Table 1, selected bond distances and angles in Table 2, and selected intermolecular distances and angles in Table 3. The molecular structure of 1 at 100 K and the atom numbering scheme are shown in Fig. (2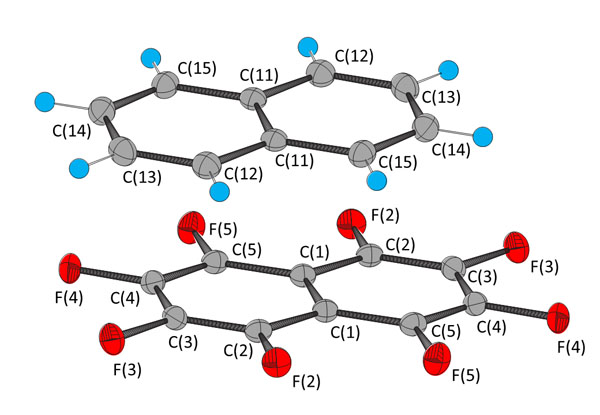 ).
).



 |
Fig. (2) The molecular structure of 1 at 100 K showing the atom numbering scheme. Thermal ellipsoids are at the 50% level. (Red = fluorine, grey = carbon, blue = hydrogen). |
The planes defined by the carbon atoms of octafluoronaphthalene and naphthalene subtend angles with the a axis of 66.7 and 69.9° respectively. The molecules of the column at the centre of the unit cell are tilted oppositely to those centred at the vertices. The planes defined by the carbon atoms of octafluoronaphthalene and naphthalene are at 34.1 and 31.5° respectively to those of analogous differently tilted molecules.
As expected, the unit cell contracts as the temperature is lowered with a greater contraction along the a and c axes, 0.110 Å (1.5%) and 0.162 Å (1.3%) respectively, than along the b axis, 0.053 Å (0.6%), on decreasing the temperature from 250 to 100 K. The coefficients of linear expansion, αL, are 1.0 × 10-4, 0.4 × 10-4 and 0.9 × 10-4 K-1 along the a, b and c axes respectively. There is a commensurate decrease in β of ca. 0.6° (by ca. 4 × 10-3 ° K-1). The values of αL are similar to those observed for 1,2,3,4-tetrafluoronaphthalene [31Bacchi, S.; Benaglia, M.; Cozzi, F.; Demartin, F.; Filippini, G.; Gavezzotti, A. X-ray diffraction and theoretical studies for the quantitative assessment of intermolecular arene-perfluoroarene stacking interactions. Chemistry, 2006, 12(13), 3538-3546.
[http://dx.doi.org/10.1002/chem.200501248] [PMID: 16506260] ] (1.0 × 10-4, 0.3 × 10-4 and 0.7 × 10-4 K-1), but the change in β, although of similar magnitude, is of opposite sign.
The bond distances and angles are invariant with temperature within experimental error, but the intermolecular distances increase with temperature (Table 3). The distance between the parallel rings is reduced by ca. 1.2% and the offset by ca. 2% on cooling from 250 to 100 K. The change in the intermolecular distance is smaller than those between other adjacent molecules (Fig. 3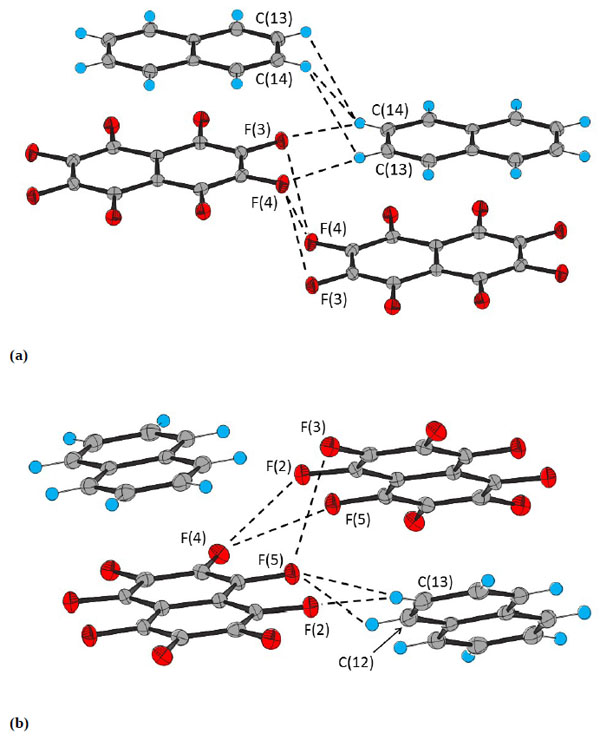 ) which are ca. 2%. The energies of the interactions were calculated for isolated pairs molecules in the gas phase by the long-range corrected functional ωB97xD [35Chai, J-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys., 2008, 10(44), 6615-6620.
) which are ca. 2%. The energies of the interactions were calculated for isolated pairs molecules in the gas phase by the long-range corrected functional ωB97xD [35Chai, J-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys., 2008, 10(44), 6615-6620.
[http://dx.doi.org/10.1039/b810189b] [PMID: 18989472] ] method using the 6-311G++(2d,2p) basis set. The values for the structure at 100 K are -59 kJ mol-1 for the π–π stacking interaction, -10, -8 and -6 kJ mol-1 for the C10F8···C10F8, C10H8···C10H8 and C10F8···C10H8 interactions shown in Fig. (3a), and -9 and -7 kJ mol-1 for the C10F8···C10F8 and C10F8···C10H8 shown in Fig. (3b). There is little variation in the energies with temperature; a decrease of < 2 kJ mol-1 on going from 100 to 250 K.
Calculations reveal that the energy of the π–π stacking interaction has a much steeper minimum than the other interactions (Fig. 4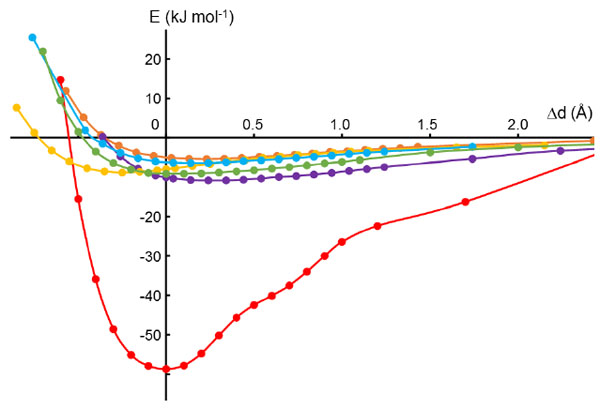 ), which are presumably van der Waals in nature. The π–π stacking interaction energy increases by ca. 8 kJ mol-1 as the distance between the two planes is increased or decreased by 0.3 Å, whereas for the other interactions the energies vary by only ca. 2 kJ mol-1 over 1 Å. Thus, the π–π stacking interaction is expected to show much less variability in geometry than the other intermolecular interactions. Experimental observation is consistent with this expectation.
), which are presumably van der Waals in nature. The π–π stacking interaction energy increases by ca. 8 kJ mol-1 as the distance between the two planes is increased or decreased by 0.3 Å, whereas for the other interactions the energies vary by only ca. 2 kJ mol-1 over 1 Å. Thus, the π–π stacking interaction is expected to show much less variability in geometry than the other intermolecular interactions. Experimental observation is consistent with this expectation.


3. MATERIALS AND METHODS
Naphthalene-octafluoronaphthalene, 1, was prepared by mixing and melting the two reagents as previously described [36Owens, A.R.; Saunders, G.C.; Thomas, H.P.; Wehr-Candler, T. Solvent-free mechanochemical syntheses and reactions of π–π stacked arene-perfluoroarene co-crystals. J. Fluor. Chem., 2015, 175, 139-144.
[http://dx.doi.org/10.1016/j.jfluchem.2015.04.004] ]. A crystal suitable for single crystal X-ray diffraction was grown from acetone. The same crystal (0.177 × 0.059 × 0.034 mm) was used to determine the structure at the four different temperatures. Diffraction data were collected on an Agilent SuperNova, single source at offset, Atlas diffractometer with graphite-monochromated Cu-Kα radiation. The structures were solved using Olex2 [37Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst., 2009, 42, 339-341.
[http://dx.doi.org/10.1107/S0021889808042726] ] structure solution programme using Charge Flipping and refined with the olex2.refine [38Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment - Olex2 dissected. Acta Crystallogr. A Found. Adv., 2015, 71(Pt 1), 59-75.
[http://dx.doi.org/10.1107/S2053273314022207] [PMID: 25537389] ] refinement package using Gauss-Newton minimization. The non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen atom positions were added in idealized positions and a riding model with fixed thermal parameters (Uij = 1.2Ueq for the atom to which they are bonded (1.5 for CH3)) was used for subsequent refinements. The function minimized was Σ[w(|Fo|2 - |Fc|2)] with reflection weights w-1 = [σ2 |Fo|2 + (g1P)2 + (g2P)] where P = [max |Fo|2 + 2|Fc|2]/3. The crystal data are presented in Table 1. CCDC 1947799 - 1947802 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viawww.ccdc. cam.ac.uk/data_request/cif.
DFT calculations using the long-range corrected functional ωB97XD [35Chai, J-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys., 2008, 10(44), 6615-6620.
[http://dx.doi.org/10.1039/b810189b] [PMID: 18989472] ] method with the 6-311G++(2d,2p) basis sets were performed using Gaussian 09 [39Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J.A., Jr; Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J.M.; Klene, M.; Knox, J.E.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Zakrzewski, V.G.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Dapprich, S.; Daniels, A.D.; Farkas, Ö.; Foresman, J.B.; Ortiz, J.V.; Cioslowski, J.; Fox, D.J. Gaussian 09, Revision A.2., 2009, ]. The energies of interaction were calculated as the difference between the energy of the species and the sum of those of its components. The C─H bonds of the experimental structures were normalized to 1.083 Å [40Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. International Tables for Crystallography., 1995, Vol. C, 685-706.] before single point energy calculations were performed.
CONCLUSION
The crystal structure of naphthalene-octafluoronaphthalene, 1, has been determined at a range of temperatures. The molecular structures are invariant with temperature. The π–π stacking between the virtually parallel naphthalene and octafluoronaphthalene molecules, which is the strongest interaction, undergoes less variation than the other intermolecular interactions, which are weaker. Calculations reveal that this is to be expected since the latter possess shallower minima.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
The authors thank the University of Waikato for support.
REFERENCES
| [1] | Hori, A. The Importance of Pi-Interactions in Crystal Engineering: Frontiers in Crystal Engineering2012. |
| [2] | Coates, G.W.; Dunn, A.R.; Henling, L.M.; Dougherty, D.A.; Grubbs, R.H. Phenyl–Perfluorophenyl Stacking Interactions: A New Strategy for Supermolecule Construction. Angew. Chem. Int. Ed. Engl., 1997, 36, 248-251. [http://dx.doi.org/10.1002/anie.199702481] |
| [3] | Dai, C.; Nguyen, P.; Marder, T.B.; Scott, A.J.; Clegg, W.; Viney, C. Control of single crystal structure and liquid crystal phase behaviour via arene–perfluoroarene interactions. Chem. Commun. (Camb.), 1999, 2493-2494. [http://dx.doi.org/10.1039/a906199a] |
| [4] | Vangala, V.R.; Nangia, A.; Lynch, V.M. Interplay of phenyl-perfluorophenyl stacking, C-H...F, C-F...π and F...F interactions in some crystalline aromatic azines. Chem. Commun. (Camb.), 2002, 12, 1304-1305. [http://dx.doi.org/10.1039/b202181a] [PMID: 12109127] |
| [5] | Smith, C.E.; Smith, P.S.; Thomas, R.L.; Robins, E.G.; Collings, J.C.; Dai, C.; Scott, A.J.; Borwick, S.; Batsanov, A.S.; Watt, S.W.; Clark, S.J.; Viney, C.; Howard, J.A.K.; Clegg, W.; Marder, T.B. Arene-perfluoroarene interactions in crystal engineering: structural preferences in polyfluorinated tolans. J. Mater. Chem., 2004, 14, 413-420. [http://dx.doi.org/10.1039/B314094F] |
| [6] | Zhu, S.; Zhu, S.; Jin, G.; Li, Z. Strong phenyl–perfluorophenyl π–π stacking and C–H⋯F–C hydrogen bonding interactions in the crystals of the corresponding aromatic aldimines. Tetrahedron Lett., 2005, 46, 2713-2716. [http://dx.doi.org/10.1016/j.tetlet.2005.01.183] |
| [7] | Serrano-Becerra, J.M.; Hernández-Ortega, S.; Morales-Morales, D. Valdés- Martínez, J. Bottom-up design and construction of a non-centrosymmetric network through π–π stacking interactions. CrystEngComm, 2009, 11, 226-228. [http://dx.doi.org/10.1039/B816630G] |
| [8] | Saunders, G.C. On the importance of π–π stacking and cation–anion interactions in the construction of non-centrosymmetric networks of bromide salts of imidazolium cations bearing arene and polyfluoroarene rings. CrystEngComm, 2011, 13, 1801-1803. [http://dx.doi.org/10.1039/c1ce00003a] |
| [9] | Itoh, T.; Kondo, M.; Kanaikea, M.; Masaoka, S. Arene–perfluoroarene interactions for crystal engineering of metal complexes: Controlled self-assembly of paddle-wheel dimers. CrystEngComm, 2013, 15, 6122-6126. [http://dx.doi.org/10.1039/c3ce40777b] |
| [10] | Lane, J.R.; Saunders, G.C.; Webb, S.J. Engineering of a polar crystal structure exclusively by π–π stacking between aryl and polyfluoroaryl groups. CrystEngComm, 2013, 15, 1293-1295. [http://dx.doi.org/10.1039/c2ce26796a] |
| [11] | Althagbi, H.I.; Lane, J.R.; Saunders, G.C.; Webb, S.J. Engineering polar crystal structures by π–π stacking of N-perfluoroarylbenzimidazoles. J. Fluor. Chem., 2014, 106, 88-95. [http://dx.doi.org/10.1016/j.jfluchem.2014.07.024] |
| [12] | Althagbi, H.I.; Edwards, A.J.; Nicholson, B.K.; Reason, D.A.; Saunders, G.C.; Sim, S.A.; van der Heijden, D.A. Arene-perfluoroarene-anion stacking and hydrogen bonding interactions in imidazolium salts for the crystal engineering of polarity. Cryst. Growth Des., 2016, 16, 174-188. [http://dx.doi.org/10.1021/acs.cgd.5b01077] |
| [13] | Oosterwijk, M.A.; Saunders, G.C.; Zou, W. Polyfluoroarene-arene π–π stacking in three directions to provide crystal polarity. J. Fluor. Chem., 2016, 188, 80-84. [http://dx.doi.org/10.1016/j.jfluchem.2016.06.015] |
| [14] | Naae, D.G. Biphenyl-perfluorobiphenyl; 1:1 molecular complex. Acta Crystallogr., 1979, B35, 2765-2768. [http://dx.doi.org/10.1107/S0567740879010499] |
| [15] | Dahl, T. Crystal structure of the 1:1 complex between N,N,N′,N′-tetramethyl-p-phenylenediamine and hexafluorobenzene. Acta Chem. Scand. A, 1979, 33a, 665-669. [http://dx.doi.org/10.3891/acta.chem.scand.33a-0665] |
| [16] | Foss, L.I.; Syed, A.; Stevens, E.D.; Klein, C.L. Structure of a naphthalene-perfluorobiphenyl complex, C10H8.C12F10. Acta Crystallogr., 1984, C40, 272-274. |
| [17] | Dahl, T. Crystal structure of the 1:1 complex between N,N,3,5-tetramethylaniline and hexafluorobenzene. Molecular-packing analysis of complexes between aromatic amines and hexafluorobenzene. Acta Chem. Scand., 1989, 43, 172-176. [http://dx.doi.org/10.3891/acta.chem.scand.43-0172] |
| [18] | Adamson, A.J.; Archambeau, Y.; Banks, R.E.; Beagley, B.; Helliwell, M.; Pritchard, R.G.; Tipping, A.E. p-Methyl-N-(pentafluorobenzylidene)aniline (1), 1,2,3,4-tetrafluoro-7-methoxyacridine (2), 1,2,3,4,7-pentafluoroacridine (3) and 3-(p-methylanilino)-1,2,4-trifluoro-7-methylacridine (4): four molecules representing key stages in the one-pot synthesis of 1,2,3,4-tetrafluoroacridines by treating pentafluorobenzaldehyde with para-substituted anilines. Acta Crystallogr., 1994, 50, 967-971. |
| [19] | Day, M.W.; Matzger, A.J.; Grubbs, R.H. 1,3,5-tris(2-Phenylethynyl)benzene tris(hexafluorobenzene)., 2001, |
| [20] | Collings, J.C.; Roscoe, K.P.; Robins, E.G.; Batsanov, A.S.; Stimson, L.M.; Howard, J.A.K.; Clark, S.J.; Marder, T.B. Arene–perfluoroarene interactions in crystal engineering 8: structures of 1:1 complexes of hexafluorobenzene with fused-ring polyaromatic hydrocarbons. New J. Chem., 2002, 26, 1740-1746. [http://dx.doi.org/10.1039/B207102A] |
| [21] | Collings, J.C.; Smith, P.S.; Yufit, D.S.; Batasnov, A.S.; Howard, J.A.K.; Marder, T.B. Arene–perfluoroarene interactions in crystal engineering. Part 10. Crystal structures of 1:1 complexes of octafluoronaphthalene with biphenyl and biphenylene. CrystEngComm, 2004, 6, 25-28. [http://dx.doi.org/10.1039/B316169B] |
| [22] | Collings, J.C.; Batsanov, A.S.; Howard, J.A.K.; Marder, T.B. Arene-perfluoroarene interactions in crystal engineering. Part 14. 1:1 Complexes of octafluoronaphthalene with fluorene and 9,10-dihydrophenanthrene. Can. J. Chem., 2006, 84, 238-242. [http://dx.doi.org/10.1139/v06-007] |
| [23] | Grzegorczyk, M.; Gdaniec, M. 2,2′,3,3′,4,4′,5,5′,6,6′-Decafluorodiphenylamine and its 1:1 cocrystal with diphenylamine. Acta Crystallogr. 2006, C62, o419-o422. (b) Stoll, I.; Brodbeck, R.; Neumann, B.; Stammler, H.-G.; Mattay, J. Controlling the self assembly of arene functionalised 2-aminopyrimidines by arene-perfluoroarene interaction and by silver(I) complex formation. CrystEngComm, 2009, 11, 306-317. |
| [24] | Dahl, T. Crystal structure of the trigonal form of the 1:1 complex between hexamethylbenzene and hexafluorobenzene. Acta Chem. Scand., 1972, 26, 1569-1575. [http://dx.doi.org/10.3891/acta.chem.scand.26-1569] |
| [25] | Dahl, T. Crystal Structure of the Triclinic Form of the 1:1 Complex between Hexamethylbenzene and Hexafluorobenzene at -40°C. Acta Chem. Scand., 1973, 27, 995-1003. [http://dx.doi.org/10.3891/acta.chem.scand.27-0995] |
| [26] | Dahl, T. Crystallographic studies of addition compounds of hexafluorobenzene. Crystal structure of the 1:1 adduct with N,N-dimethylaniline. Acta Crystallogr., 1977, B33, 3021-3024. [http://dx.doi.org/10.1107/S0567740877010188] |
| [27] | Dahl, T. Structure from a twinned crystal of the triclinic form of the 1:1 complex between N,N-dimethylaniline and hexafluorobenzene [C8H11N.C6F6] at 120 K. Acta Crystallogr., 1985, C41, 931-933. |
| [28] | Overell, J.S.W.; Pawley, G.S. An X-ray single-crystal study of the molecular system C6F6.C6D6. Acta Crystallogr., 1982, B38, 1966-1972. [http://dx.doi.org/10.1107/S0567740882007687] |
| [29] | Williams, J.H.; Cockcroft, J.K.; Fitch, A.N. Structure of the lowest temperature phase of the solid benzene–hexafluorobenzene adduct. Angew. Chem. Int. Ed. Engl., 1992, 31, 1655-1657. [http://dx.doi.org/10.1002/anie.199216551] |
| [30] | Gung, B.W.; Amicangelo, J.C. Substituent effects in C6F6-C6H5X stacking interactions. J. Org. Chem., 2006, 71(25), 9261-9270. [http://dx.doi.org/10.1021/jo061235h] [PMID: 17137351] |
| [31] | Bacchi, S.; Benaglia, M.; Cozzi, F.; Demartin, F.; Filippini, G.; Gavezzotti, A. X-ray diffraction and theoretical studies for the quantitative assessment of intermolecular arene-perfluoroarene stacking interactions. Chemistry, 2006, 12(13), 3538-3546. [http://dx.doi.org/10.1002/chem.200501248] [PMID: 16506260] |
| [32] | Potenza, J.; Mastropaolo, D. Naphthalene-octafluoronaphthalene, 1:1 Solid Compound. Acta Crystallogr., 1975, B31, 2527-2529. [http://dx.doi.org/10.1107/S0567740875008011] |
| [33] | Bagryanskaya, I.Y.; Gatilov, Y.V.; Maksimov, A.M.; Platonov, V.E.; Zibarev, A.V. Supramolecular synthons in crystals of partially fluorinated fused aromatics: 1,2,3,4-Tetrafluoronaphthalene and its aza-analogue 1,3,4-trifluoroisoquinoline. J. Fluor. Chem., 2005, 126, 1281-1287. [http://dx.doi.org/10.1016/j.jfluchem.2005.06.011] |
| [34] | Cozzi, F.; Bacchi, S.; Filippini, G.; Pilati, T.; Gavezzotti, A. Synthesis, X-ray diffraction and computational study of the crystal packing of polycyclic hydrocarbons featuring aromatic and perfluoroaromatic rings condensed in the same molecule: 1,2,3,4-tetrafluoronaphthalene, -anthracene and -phenanthrene. Chemistry, 2007, 13(25), 7177-7184. [http://dx.doi.org/10.1002/chem.200700267] [PMID: 17568459] |
| [35] | Chai, J-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys., 2008, 10(44), 6615-6620. [http://dx.doi.org/10.1039/b810189b] [PMID: 18989472] |
| [36] | Owens, A.R.; Saunders, G.C.; Thomas, H.P.; Wehr-Candler, T. Solvent-free mechanochemical syntheses and reactions of π–π stacked arene-perfluoroarene co-crystals. J. Fluor. Chem., 2015, 175, 139-144. [http://dx.doi.org/10.1016/j.jfluchem.2015.04.004] |
| [37] | Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst., 2009, 42, 339-341. [http://dx.doi.org/10.1107/S0021889808042726] |
| [38] | Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment - Olex2 dissected. Acta Crystallogr. A Found. Adv., 2015, 71(Pt 1), 59-75. [http://dx.doi.org/10.1107/S2053273314022207] [PMID: 25537389] |
| [39] | Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J.A., Jr; Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J.M.; Klene, M.; Knox, J.E.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Zakrzewski, V.G.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Dapprich, S.; Daniels, A.D.; Farkas, Ö.; Foresman, J.B.; Ortiz, J.V.; Cioslowski, J.; Fox, D.J. Gaussian 09, Revision A.2., 2009, |
| [40] | Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. International Tables for Crystallography., 1995, Vol. C, 685-706. |