- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Systems Biology Journal
(Discontinued)
ISSN: 1876-3928 ― Volume 5, 2014
Lytic Efficiency of Mycobacteriophages
Sameer Hassan, Azger Dusthackeer, Balaji Subramanyam, C Ponnuraja, Gomathi N Sivaramakrishnan , Vanaja Kumar*
Abstract
Mycobacteriophages D29 and TM4 are the two virulent phages widely used for the study of mycobacterial genetics. Both the phages are capable of killing Mycobacterium tuberculosis but the efficiency of these phages in killing has not been evaluated and compared. There are reports based on codon usage analysis where TM4 is predicted to be a better killing phage over D29 which corroborated with the whole genome in silico analysis. In addition a kill assay using 5 wild type virulent mycobacteriophages viz. D29, TM4, I3, Che7 and Che11 was performed to study the killing efficiency of these phages using LRP assay. Based on the results, D29 was found to infect all the 10 clinical strains of M. tuberculosis and significantly reduced RLU at 3 hours and this effect continued up to 24 hours. Thus, D29 is found to have better killing efficiency than TM4 contradicting the earlier predictions. In silico analysis of holin and lysin genes of TM4 and D29 substantiated our findings.
Article Information
Identifiers and Pagination:
Year: 2010Volume: 3
First Page: 21
Last Page: 28
Publisher Id: TOSYSBJ-3-21
DOI: 10.2174/1876392801003010021
Article History:
Received Date: 31/8/2009Revision Received Date: 11/11/2009
Acceptance Date: 1/2/2010
Electronic publication date: 8/5/2010
Collection year: 2010
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http: //creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Department of Bacteriology, Tuberculosis Research Centre, Chetput, Chennai-600 031, India; Tel: +91-44-28369659; Fax: +91-44-28362528; E-mail: vanajakumar@trcchennai.in
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 31-8-2009 |
Original Manuscript | Lytic Efficiency of Mycobacteriophages | |
INTRODUCTION
The complex cell wall architecture of Mycobacterium tuberculosis defies the use of molecular tools in mycobacterial research. The discovery of mycobacteriophages, have facilitated the development of mycobacterial genetic systems and provided insights into viral diversity and the evolutionary mechanisms that generate them [1-5]. The resurgence of tuberculosis and emergence of drug resistance has stimulated fresh research on mycobacteriophages.
Genome sequences of 61 mycobacteriophages are available on online databases. The availability of these sequences has helped in understanding the evolutionary relationship and genetic diversity among these phages. The comparative genome analysis of 14 mycobacteriophages revealed that they are relatively large in genome size and contain large number of previously unidentified genes and are highly diverse at both the nucleotide and amino acid sequence levels [1]. The diversity at the nucleotide sequence level of mycobacteriophages appears to be greater than that of the diary phages [6] or staphylococcal phages [7]. Recently, Hatfull et al., has grouped 3357 proteins of 30 mycobacteriophage genomes into 1536 phamilies of related sequences that represent 50% of total number of phamilies in the mycobacteriophage population [8].
Mycobacteriophage TM4 is a virulent tailed-phage with double stranded DNA [9]. Its genome is approximately 50 kb in length and contains cohesive termini [9, 10]. Mycobacteriophage D29 is a virulent phage, capable of infecting both fast and slow growing mycobacteria. It is a close relative of mycobacteriophage L5 with a 3.6 kb deletion removing the repressor gene that accounts for the inability of D29 to form lysogens [11]. TM4, D29 along with L5 and BxZ2 are identified to have the broadest host range capable of forming plaques on all of the slow growing species [12]. The ability of D29 to enter and kill macrophages infected with M. tuberculosis was reported earlier whereas TM4 cannot enter macrophage on its own due to lack of specific receptors. D29 genome might encode specific protein receptors which are recognized by the macrophage facilitating the phagocytosis of D29 [13].
Study of codon usage in fourteen mycobacteriophage genomes revealed the role of mutational pressure at the third synonymous codon position and translation selection acting in highly expressed genes [14]. Based on another study on codon usage of six lytic mycobacteriophage genomes, the role of mutational and translational forces acting on these genomes were determined [15]. The authors further identified that Che9c, BxZ1 and TM4 may be extremely virulent phages as majority of their genes have high translation efficiency.
In the current study, we have analyzed the killing efficiency of five lytic mycobacteriophages using in vitro methodology and further corroborated with bioinformatics analysis.
MATERIAL AND METHODS
Sequence
The protein coding sequences of D29 and TM4 were downloaded from GenBank. Genes were selected based on the following criteria: (a) genes having ≥ 300 bases (b) genes having proper start and stop codons (c) genes with no stop codons within.
Bioinformatics
- Sequence analysis: PHI- Blast (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used for database search. TMHMM and SignalP were used for the prediction of transmembrane helices and signal peptide respectively. Plotting of hydrophobicity profile was done with Bioedit software. ClustalW was used for multiple sequence alignment while PFAM and InterPro were made use of for domain analysis.
- Codon usage analysis was performed for the protein coding sequences of TM4 and D29. The relative synonymous codon usage (RSCU) in both phages was determined to study the overall codon usage variation among the genes and genomes. RSCU is defined as the ratio of the observed frequency of codons to the expected frequency if all the synonymous codons for those amino acids are used equally [16]. RSCU values greater than 1.0 indicates that the corresponding codons are more frequently used than expected and vice versa. GC3S is the frequency of (G+C) and A3S, T3S, G3S, and C3S are the frequencies of A, T, G, and C at the synonymous third positions of codon. These frequencies were calculated for the genes of both the genomes. Nc, the effective number of codons used by a gene is generally used to measure the bias of synonymous codons and it is independent of amino acid compositions and codon number [17]. The values of Nc range from 20 (when one codon is used per amino acid) to 61 (when all the codons are used with equal probability). Highly biased genes are generally highly expressed [18]. As there is no information of gene expression level of bacteriophages, we have considered the highly biased genes as highly expressed in the case of mycobacteriophages under study. Hence the sequences in which the Nc values were less than 35 and those having Nc values greater than 55 were considered to be highly and lowly expressed respectively. Correspondence analysis using RSCU values were performed. CodonW 1.3 (http://www.molbiol.ox.ac.uk/cu) was used for correspondence analysis [19] and for analyzing the above mentioned parameters.
Phages and Bacteria
A total of ten clinical strains of Mycobacterium tuberculosis isolated at Tuberculosis Research Centre, Chennai were grown at 37°C on Lowenstein-Jensen (LJ) slopes. Suspensions of these strains equivalent to McFarland’s standard # 2 were prepared in Middlebrook 7H9 (Difco) supplemented with Bovine Albumin-Dextrose complex and 0.5% glycerol. Each suspension was divided into 6 aliquots of 1.5 ml each to which 300 µl of phages namely D29, TM4, I3, Che7, Che11 at a titer of 1x 1010 pfu/ml was added. Mycobacteriophage buffer was used as a negative control. After 3 and 24 hours of incubation, the number of tubercle bacilli remaining alive was determined by performing a luciferase reporter phage assay. This was done by adding 50 µl of luciferase reporter phage phAE129 (1x 1010 pfu/ml) to 250 µl of initial wild type phage-cell mixture and incubating for 4 hours. Light output was measured as relative light units (RLU) using the luminometer (Monolight 2010) with 10 seconds integration time after adding 0.33mM D-luciferin (R&D Systems, Minneapolis) in 0.05M sodium citrate buffer (pH 4.5) to phage–cell mixture. Reduction in RLU by 50% compared to the buffer control was considered to reflect significant reduction in viability.
RESULTS
Gene Organization
The genome of D29 and TM4 is 49 kb and 52 kb long respectively. Both the genomes have two lysin and one holin coding genes in their genomes. Double stranded DNA bacteriophages use the dual system i.e endolysin–holin to achieve lysis of their bacterial host. TM4 and D29 being lytic mycobacteriophages possess the dual lytic system to lyse the host cell. Comparison of the organization of endolysin-holin genes in TM4 and D29 genomes (Fig. 1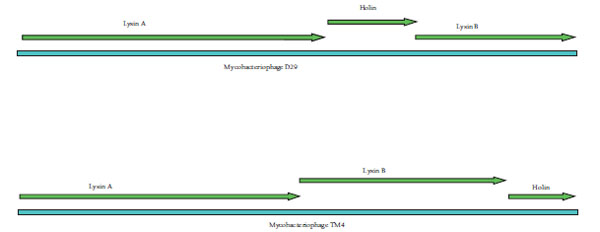 ) revealed holin gene being sandwiched between two lysin genes in D29 whereas in TM4, holin was found to be present at the end of the two lysin genes.
) revealed holin gene being sandwiched between two lysin genes in D29 whereas in TM4, holin was found to be present at the end of the two lysin genes.
 |
Fig. (1) Organization of lysin and holin genes in the genome of D29 and TM4. |
Codon Usage Analysis
The overall GC content of TM4 and D29 was found to be 68.7 and 63.78 respectively. As both the phages carry GC-rich genomes, codons ending in G and C should be predominant in the coding sequences. It was observed that the Nc values for TM4 and D29 ranged from 27.79 to 44.42 and 29.38 to 59.2 respectively, whereas the GC3s values ranged from 71.2 to 95.3 and 63.5 to 93.3 respectively. This showed high variation in the Nc and GC3s values among the genes of both the genomes.
Nc plots (a plot of Nc versus GC3s) and correspondence analysis (CA) determine the determinants of the codon usage variations in different organisms. The Nc plot for the genes of both TM4 and D29 phages showed that majority of the genes lay below the expected curve towards GC rich regions suggesting that these phages displayed an additional codon usage bias which was independent of GC3s (Fig. 2 ).
).
 |
Fig. (2) Nc plot of genes from D29 (green) and TM4 (black). |
Apart from mutational bias, translational bias also plays a major role in shaping the codon usage of these phages. A scatter plot was drawn between the position of genes along the first major axis and Nc values (Fig. 3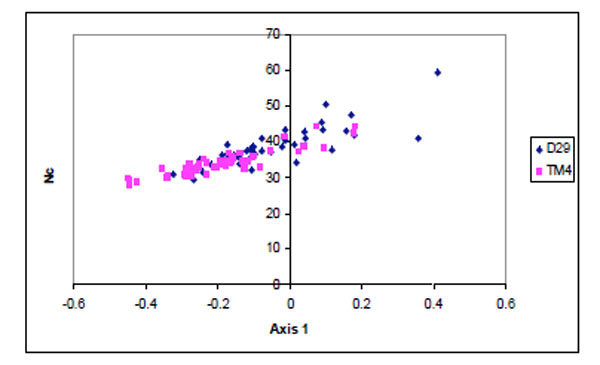 ). Majority of the TM4 genes were found to have lower Nc values than D29.
). Majority of the TM4 genes were found to have lower Nc values than D29.
 |
Fig. (3) Scatter plot of the genes of D29 (blue) and TM4 (pink)against Nc values. |
Correspondence analysis showed the distribution of the genes of the two genomes along the first two major axes, accounting for 12.1 % and 8.3 % respectively of the total variation. Based on the results obtained (data not shown) there were 5 over-represented codons in the genes located at the negative side of the first major axis. Out of 5 predominant codons, 4 ended in C and the other ended in G at the third position.
A comparative analysis of the positions of genes along first major axis and the nucleotide composition at the third codon position revealed that the first axis was positively correlated with A3 and T3 but negatively correlated with G3 and C3 (Table 1). The results suggested that A and T ending codons were clustered on the positive side whereas G and C ending codons were located on the negative side of first major axis.

Kill Assay
Among the five phages tested D29 was found to be most effective in producing more than 50% killing in all the ten strains at 3 and 24 hours (Fig. 4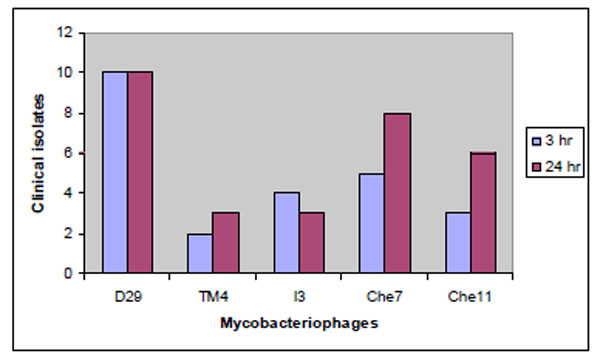 ). Che7 exhibited similar efficiency in 8 strains at 24 hours, whereas TM4 was the least effective among the phages tested. It was able to produce more than 50% killing in only 3 of the clinical isolates.
). Che7 exhibited similar efficiency in 8 strains at 24 hours, whereas TM4 was the least effective among the phages tested. It was able to produce more than 50% killing in only 3 of the clinical isolates.
 |
Fig. (4) Lytic activity of phages in clinical strains of Mycobacterium tuberculosis. |
Sequence Analysis of Holin and Lysin
Nc Values for Lysin and Holin
Apart from overall Nc values for both the genomes, Nc values for lysin and holin genes were compared separately. Based on the Nc values (Table 2) of lysin and holin of both genomes, the lysin gene of TM4 showed lower Nc value than that of D29, whereas holin gene of D29 showed lower Nc value than of TM4.

Sequence Analysis of Lysin and Holin of TM4
Gp29 and Gp30 of TM4 are identified to code for lysin protein and are 547 and 400 amino acid long respectively. Based on the prediction of phage genes into gene families by Hatfull et al. for 30 mycobacteriophage genome, Pham7 contains Lysin A genes and Pham9 contains Lysin B genes [6]. In the current study GP29 of TM4 was identified as Lysin A and GP 30 as Lysin B as predicted earlier.
Lysin A protein of TM4 was recognized to have N-acetylmuramoyl-L-alanine amidase domain (209-356) based on PFAM analysis. Blast search using Lysin A protein of TM4 identified significant similarity to N-acetylmuramoyl-L-alanine amidase protein encoded by Mycobacterium sp. MCS (58 % with aa). GP29 of TM4 was therefore designated as N-acetylmuramoyl-L-alanine amidase. Multiple sequence alignment of GP29 with other known amidase proteins revealed the conservity of zinc binding and catalytic triad residues (Fig. 5 ).
).

Lysin B protein encoded by GP30 was identified to have a peptidoglycan binding domain at the N-terminal region of GP30. Blast search and PHYRE using Lysin B of TM4 as query sequence identified similarity to α/β hydrolases. Multiple alignment of Lysin B sequences of mycobacteriophages identified the presence of a conserved pentapeptide Gly-Tyr-Ser-Gln-Gly motif at positions 162–166 of the Lysin B amino acid sequence matching with the characteristic Gly-X-Ser-X-Gly motif found in lipolytic enzymes (Fig. 6 ).
).

GP31 of TM4 codes for holin protein of 128 amino acids consisting of two transmembrane helices with highly charged N terminal and C terminal (Fig. 7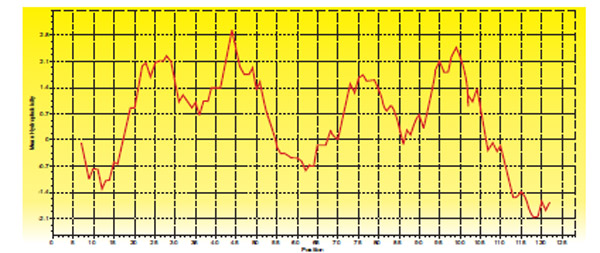 ).
).
 |
Fig. (7) Hydropathy plot of Holin protein of TM4 |
Sequence Analysis of Lysin and Holin of D29
GP 10 and GP 12 of D29 codes for Lysin A (493 aa) and Lysin B (254 aa) respectively. Lysin A was identified to have Lysozyme like domain (198-362). The Gly-X-Ser-X-Gly motif found in lipolytic enzymes along with the residues involved in the catalytic triad was identified to be well conserved in GP 12 (lysin B) protein sequence of D29 (Fig. 6).
GP11 of D29 codes for holin protein belonging to Class II holin. Two transmembrane helices were predicted using TMHMM. GP11 was found to have a short charged N terminal region and a highly charged C terminal region (Fig. 8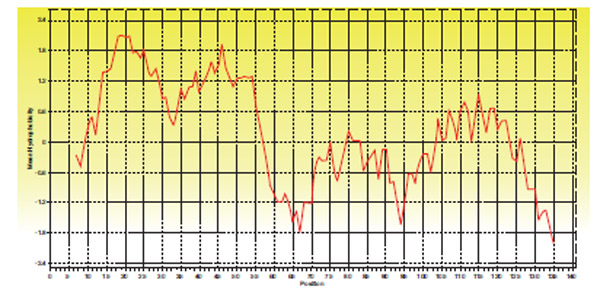 ).
).
 |
Fig. (8) Hydropathy plot of Holin protein of D29 |
DISCUSSION
In the current study, the codon usage of D29 and TM4 genomes and Nc values for holin and lysin genes coded by both genomes were analysed and compared. Based on the Nc plot, apart from the mutational force, some other factors are also understood to play an important role in shaping the codon usage in both the genomes. Identification of significant positive correlation between Nc values and Axis 1 revealed that substantial number of TM4 genes had lower Nc values as compared to D29 genes. On the basis of this result it is predicted that a balance between mutational force and selection due to translational efficiency is strongly operating in selecting the codon usage variation among the genes of these phages. The number of highly biased genes were more in TM4 (72%) than in D29 (30%) genome. Several reports have shown earlier that synonymous codon usage in the highly expressed genes of diverse array of organisms is influenced by cellular tRNA abundance [20-25]. Based on the comparative analysis between M. tuberculosis H37Rv tRNA and synonymous codon usage, it is identified that all the over represented codons of D29 and TM4 are recognized by host tRNA. On the basis of our analysis it is expected that more number of TM4 genes should be efficiently translated leading to better lytic activity and faster killing. Earlier report by Ranjan et al. [15] predicting the genes of TM4 to be expressed rapidly by the host‘s translation machinery corroborated with our finding.
The better translational efficiency of TM4 was expected to quicken the lysis process qualifying it to be a better lytic phage. However, the lytic performance of TM4 was poor in all the ten strains tested in vitro whereas D29 was efficient in effectively killing all of them at 3 hours and that effect was observed to continue up to 24 hours (Fig. 4).
Subsequently, holin and lysin genes of both the phages were analysed in detail to understand the performance of D29 over TM4 on the clinical strains of M. tuberculosis. Holin sequences of TM4 (Class I holin) and D29 (Class II holin) were found to be highly divergent sharing no amino acid similarity. Holin of TM4 was identified to be longer possessing a highly charged N terminal region whereas holin of D29 was shorter possessing a lowly charged N terminal region. Presence of highly charged N-terminal regions in holins has been reported to be a typical feature of holin-inhibitors [26]. Thus, it is likely that the presence of the highly charged N terminal region of holin in TM4 acted as antiholin thereby delaying the lysis process of TM4.
Virulent phages contain more tRNAs than temperate ones. They have higher codon usage biases and compositional differences with respect to the host genome. Even though phages use most of the cell’s translation machinery, they can complement it with their own genetic information to attain higher fitness [27]. The presence and expression of phage tRNA genes that are already present in the host genome will have additional benefit on the phage genome [27]. So the presence of 5 tRNA genes in D29 genome compared to none in TM4 should further increase the fitness of translational efficiency of the genes of D29, allowing the expression of low biased genes in the genome. Moreover among the over-represented codons of TM4 phage few were recognized by the abundant host tRNAs compared to D29.
The cell wall of mycobacteria has a high lipid content which is absent in other gram positive bacteria. Mycobacterial cell envelop consists of long-chain mycolic acids esterified to an arabinogalactan polysaccharide, which is attached to the peptidoglycan backbone. This mycolyl-arabinogalactan-peptidoglycan complex (core) intercalates with an array of unusual free lipids, resulting in an effective external permeability barrier [28, 29]. The presence of additional lysis genes such as Rz and Rz1 was reported earlier in bacteriophages enabling the efficient infection of Gram negative bacteria [30, 31]. Similarly, the presence of mycolic acid in mycobacterial cell wall may require additional lysis genes in order to dislocate the complex mycolic acid. The lysin coding genes of mycobacteriophages had been organized into phamilies such as Pham 7 and Pham 9 of related sequences [8]. Pham 7 contains lysin A genes, one of the two lysins of mycobacteriophages and Pham 9 contains lysin B genes. Lysin B in the genomes of D29 and TM4 were identified to have the conserved Gly-X-Ser-X-Gly motif of lipolytic enzyme. Based on Blast analysis, it was found that Lysin B gene is present exclusively in mycobacteriophage genomes probably enabling the infection of M. tuberculosis effectively.
Multiple sequence alignment of Lysin B of mycobacteriophages revealed the conservity of Gly-X-Ser-X-Gly motif characteristic of lipolytic enzymes in 19 of them (Fig. 6). However the amino acid sequence of Lysin B did not show characteristic features of any of the lipase families identified by Arpigny & Jaeger [32-33]. Based on profile-profile matching algorithm of PHYRE, lysin B was identified to show similarity to cutinase like family of proteins. An important feature of the α/β hydrolase superfamily is the presence of Asp and His residues which form the catalytic triad together with the Ser residue [32, 34-35]. LysB protein of Ms6 mycobacteriophage was reported to belong to the family of serine hydrolases [31] as is the case for lipid hydrolases [34]. The alignment (Fig. 6) showed that Asp and His residues were conserved among the homologous proteins of the mycobacteriophages. Although three Asp residues (Asp129, Asp166, Asp224) of D29 lysin B were totally conserved, Asp129 together with His163 might be good candidates for the catalytic triad, obeying the order Ser- Asp-His. However, in GP30 of TM4, Asp237 (Asp166 in D29) was conserved but Asp129 and His 163 in D29 are replaced by Gly and Ala in TM4 respectively. These amino acid substitutions may probably influence the activity of Lysin B in TM4 which is not yet experimentally proved.
Though over all codon bias of TM4 genome was stronger than that of D29, the details of lysis cassettes and presence of tRNAs of both the phages revealed that D29 was well equipped to efficiently lyse the host cells faster. The killing efficiency of D29 compared to four other lytic phages including TM4, as observed in the kill assay resulted in loss of viability of M. tuberculosis clinical strains as early as 3 hours. Thus, predictions by bioinformatics approach and laboratory experiments mutually contributed to each other quickening the understanding of the biological processes.
ACKNOWLEDGEMENTS
This work was financially supported by ICMR-Biomedical Informatics Centre. We also acknowledge K. Ramakrishnan and G. Vadivu for their technical assistance.