- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Cell Signaling Journal
(Discontinued)
ISSN: 1876-3901 ― Volume 4, 2012
‘Chromosomal Rainbows’ Detect Oncogenic Rearrangements of Signaling Molecules in Thyroid Tumors
Benjamin O'Brien*, 1, 2, 3, Gregg H. Jossart1, 4, 6, Yuko Ito1, 7, Karin M. Greulich-Bode1, 8, Jingly F. Weier1, 5, Santiago Munne9, Orlo H. Clark4, Heinz-Ulrich G. Weier1
Abstract
Altered signal transduction can be considered a hallmark of many solid tumors. In thyroid cancers the receptor tyrosine kinase (rtk) genes NTRK1 (Online Mendelian Inheritance in Man = OMIM *191315, also known as ‘TRKA’), RET (‘Rearranged during Transfection protooncogene’, OMIM *164761) and MET (OMIM *164860) have been reported as activated, rearranged or overexpressed. In many cases, a combination of cytogenetic and molecular techniques allows elucidation of cellular changes that initiate tumor development and progression. While the mechanisms leading to overexpression of the rtk MET gene remain largely unknown, a variety of chromosomal rearrangements of the RET or NTKR1 gene could be demonstrated in thyroid cancer. Abnormal expressions in these tumors seem to follow a similar pattern: the rearrangement translocates the 3’- end of the rtk gene including the entire catalytic domain to an expressed gene leading to a chimeric RNA and protein with kinase activity. Our research was prompted by an increasing number of reports describing translocations involving ret and previously unknown translocation partners.
We developed a high resolution technique based on fluorescence in situ hybridization (FISH) to allow rapid screening for cytogenetic rearrangements which complements conventional chromosome banding analysis. Our technique applies simultaneous hybridization of numerous probes labeled with different reporter molecules which are distributed along the target chromosome allowing the detection of cytogenetic changes at near megabasepair (Mbp) resolution. Here, we report our results using a probe set specific for human chromosome 10, which is altered in a significant portion of human thyroid cancers (TC’s). While rendering accurate information about the cytogenetic location of rearranged elements, our multilocus, multi-color analysis was developed primarily to overcome limitations of whole chromosome painting (WCP) and chromosome banding techniques for fine mapping of breakpoints in papillary thyroid cancer (PTC).
Article Information
Identifiers and Pagination:
Year: 2010Volume: 2
First Page: 13
Last Page: 22
Publisher Id: TOCELLSJ-2-13
DOI: 10.2174/1876390101002010013
Article History:
Received Date: 02/05/2010Revision Received Date: 21/08/2010
Acceptance Date: 22/10/2010
Electronic publication date: 15/12/2010
Collection year: 2010
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http: //creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Department of Cardiothoracic Anaesthesia and Intensive Care, Imperial College Healthcare NHS Trust, Hammersmith Hospital, London W12 0HS, UK; Tel: +44-2033133991; Fax: +44-2033135373; E-mail: benobrien@doctors.org.uk
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 02-05-2010 |
Original Manuscript | ‘Chromosomal Rainbows’ Detect Oncogenic Rearrangements of Signaling Molecules in Thyroid Tumors | |
INTRODUCTION
The transformation of normal human to neoplastic cells is often accompanied by visible cytogenetic alterations, i.e., chromosomal rearrangements such as translocations, deletions or gene amplifications, leading to the deletion or loss of function of tumor suppressing genes or activation of tumor promoting genes. Abnormal signal transduction secondary to activation of proto-oncogenes by fusion to expressed heterologous genes is a common mechanism of malignant transformation in solid tumors [1].
Increasingly, structural chromosome aberrations are found to be pathognomonic for certain solid tumors.
In PTC [2], it has been reported that activation of rtk genes such as RET or neurotrophic receptor kinase type I (NTRK1) by intra- or interchromosomal rearrangements causes altered signal transduction and ultimately malignant transformation of the cells [3-9]. Since the key events in neoplastic transformation are expected to be among the most commonly found alterations in phenotypically related tumors, the localization of chromosomal breakpoints is an important step towards positional cloning of the disease causing gene(s). Towards this end, numerous techniques have been developed, several of which depend on enumeration and localization of specific DNA sequences in situ.
Chromosome banding techniques require high quality metaphases that are often difficult to obtain from tumor cells. Additionally, the ability to detect subtle rearrangements depends not only on the quality of the metaphase spreads but also on the experience of the cytogeneticist performing the study. The development of FISH with whole chromosome painting (WCP) probes was a major advancement over classical cytogenetic banding techniques [10, 11]. Screening for translocations involving non-homologous chromosomes by multi-color WCP has proven very useful [12-15]. Nevertheless, WCP probes are unsuitable to detect most intrachromosomal rearrangements and small deletions [1, 16].
Physically mapped large insert DNA clones and access to recombinant DNA libraries representing many equivalents of the human genome now allow the development of molecular cytogenetic procedures that complement conventional cytogenetic analysis techniques, i.e., chromosome banding, and will eventually lead to an accelerated identification of disease genes. Furthermore, advances in computer-assisted multi-color fluorescence microscopy have aided the development of high resolution, artificial color banding patterns for each human chromosome based on hybridization of specifically coded DNA probes. This strategy has been suggested earlier and the pioneering work of several groups deserves appropriate credit.
Lichter and coworkers [17] demonstrated how data from genetic and physical maps of chromosome 10 can be linked by in situ hybridization on to metaphase chromosome spreads and that selecting clones in adequately large intervals ensures consistent probe order in FISH experiments. Concurrently, Lengauer, et al. proposed the generation of what they called "Chromosomal Bar Codes" by combining WCP’s with locus-specific probes derived from yeast artificial chromosome (YAC) clones. Although their contribution to the field was significant, the actual resolution of their 'barcodes' suffered from having only relatively few probes spread over 24 different chromosome types and a limitation to only two haptens and, thus, two probe fluorescence colors [11]. Combinatorial probe labeling [18-20] with three independent fluorescence wavelength intervals for probe detection allows one to significantly increase the density of probes that can be hybridized simultaneously along the target chromosome thereby approaching the high resolution of banding techniques. Liehr, et al. described 'Multicolor Chromosomal Banding' utilizing combination of YAC/BAC probes and region-specific microdissection DNA libraries for chromosomes 2, 13, and 22, which yields a resolution in the order 4-10 clones per chromosome band or roughly 2-3 million base pairs (Mbp) [21]. More recently, Lu, et al. described 'DNA probe pooling' as a way to improve hybridization efficiency and speed [22] emphasizing the fact that larger FISH targets lead to consistently higher FISH success.
Our main interest in developing the technique described here stems from our ongoing investigations of genetic alterations in human thyroid carcinomas. In these tumors, certain chromosomes (1, 2, 3, 10, 11, 17, and 19) appear to be more frequently rearranged than others [23-29] prompting us to develop probe sets which bind specifically to selected sites along these chromosomes. To obtain the power necessary to detect small deletions and rearrangements reported for thyroid carcinomas [3, 30-32], we chose probes which map along the target chromosome(s) in intervals of roughly 0.06 units of its fractional length [17]. For a C-group chromosome such as chromosome 10 with an estimated size of ~140 Mbp, this number translates to approximately 8 Mbp between hybridization probes. Thus, for covering chromo-some 10 with a multicolor 'chromosomal rainbow' set (CR10) we selected 16 clones from the Dupont P1 and the CEPH/Genethon yeast artificial chromosome (YAC) libraries [33, 34]. In this article, we describe the selection of the 12 P1 and four YAC clones, and the evaluation and application of CR10. After optimization using normal metaphase spreads, the chromosomal rainbow technique was applied to metaphase spreads of the cell line TPC-1 established from a papillary variant of TC [30]. As reported earlier, this cell line carries a complex t(1;10;21) translocation, a RET-activating RET/PTC1 rearrangement and a deletion involving the locus D10S170 [8, 32, 35, 36] (Fig. 1A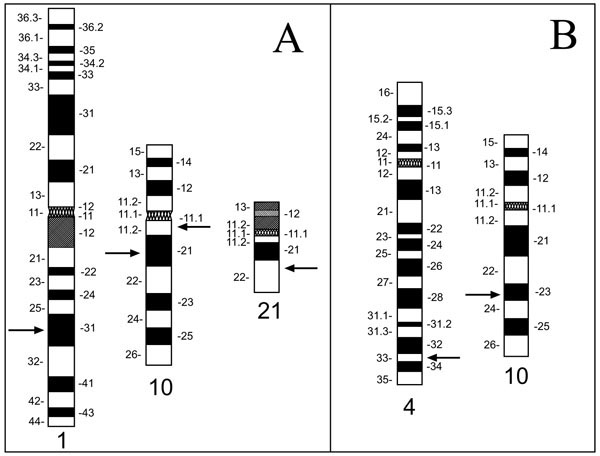 ). We furthermore applied the CR10 set in two typical applications: characterization of a marker chromosome in the follicular TC cell line FTC-236, derived from a pulmonary metastases, and for breakpoint mapping in the case of a balanced reciprocal translocation t(4;10) in a patient enrolled in an in vitro fertilization (IVF) program (Fig. 1B) [21, 37, 38].
). We furthermore applied the CR10 set in two typical applications: characterization of a marker chromosome in the follicular TC cell line FTC-236, derived from a pulmonary metastases, and for breakpoint mapping in the case of a balanced reciprocal translocation t(4;10) in a patient enrolled in an in vitro fertilization (IVF) program (Fig. 1B) [21, 37, 38].

MATERIALS AND METHODS
Cells and Metaphase Spreads
Metaphase spreads were prepared from established cell lines and PHA stimulated short term lymphocyte cultures using standard techniques of colcemid block, hypotonic treatment with 75 mM KCl and fixation in acetic acid:methanol (1:3, vol.: vol.) [21]. Metaphase spreads of the thyroid cancer cell lines TPC-1 and FTC-236 were prepared from cells grown on slides [39]. Slides were stored at -20°C under nitrogen in sealed plastic bags. Prior to hybridization, cells on slides underwent digestion with RNAse A (Roche Molecular Systems, Indianapolis, IN) (1 mg/ml at 37°C for 1 hour) and blocking with gelatin (0.05% in water, weight:vol.) (Sigma, St. Louis, MO) [40].
Probe DNA Preparation and Labeling
The P1 DNAs were extracted from overnight cultures following an alkaline lysis protocol [41]. The YAC DNAs were isolated from 48 hour liquid cultures following a standard protocol using yeast lytic enzyme (ICN, Aurora, OH) [42]. The DNAs were quantitated by Hoechst fluorometry using a Hoefer TK 100 instrument (Hoefer, South San Francisco). Probe DNAs were labeled by random priming as described previously [35, 42]. Probes used in the CR10 rainbow set are listed in Table 1. Our probe labeling and detection scheme is summarized in Table 2. Yellow fluorescence signals were obtained using probes that had been random primed in the presence of equimolar ratios of digoxigenin-11- (dig) and fluorescein-12-dUTP (fluorescein isothyocyanate, FITC; Roche).

Fluorescence In Situ Hybridization
The hybridization mixtures contained 70 µl of human COT-1™ DNA (1 mg/ml, Gibco/LTI), 17 µl of salmon sperm DNA (10 mg/ml; Invitrogen. Carlsbad, CA) and between 60 - 120 ng of each probe, depending on hybridization quality and signal intensity of individual probes. The DNA was precipitated in ethanol and resuspended in 3 µl water, before 7 µl of Master Mix 2.1 [42] were added in order to have the probe finally dissolved in 55% formamide (FA; Invitrogen), 10% dextran sulfate and 2xSSC, pH 7.0. The probe mixture was then heat-denatured at 75°C for 7 minutes and preannealed at 37°C for 30 min. Slides were denatured for 3.5 minutes in 70% FA/2x SSC, pH 7.0 at 75°C and dehydrated in a 70%, 85%, 100% ethanol series, two minutes each. About 10 µl of hybridization mixture were placed on the denatured samples, covered with a 22 mm x 22 mm coverslip, sealed with rubber cement and incubated for 42-46 hours at 37°C in a humidity chamber.
Detection
Posthybridization washes were performed as described and included three washes in PN buffer (Na2HPO4-7 hydrate/NaH2PO4-1 hydrate, pH 8, 0.05% Non-Idet P-40 (Sigma)) [41, 42]. Unspecific binding sites were blocked with PNM (5% nonfat dry milk, 0.1% NaN3 in PN buffer) [43]. The different layers of fluorochrome-labeled avidin or antibodies were applied in the concentrations listed in Table 3. The yellow color was generated by overlapping green and red signal, i.e., by hybridization of probes labeled simultan-eously with digoxigenin and FITC. After each antibody layer, slides underwent three 10 min. washes in PN with agitation and were again blocked with PNM. After the final wash in PN, slides were washed 10 min. in 0.1% Tween-20 (Sigma) in 2xSSC, mounted with 4,6-diamino-2-phenylin-dole (DAPI) (0.1µg/ml in (0.1% p-phenylenediamine dihydrochloride (Sigma), 0.1x PBS (Invitrogen), 45 mM NaHCO3, 82% glycerol, pH 8.0) and coverslipped. Images of the in situ hybridization results were recorded on a Zeiss Axioskope microscope (Zeiss, Oberkochen, Germany) equipped with a cooled CCD camera (Photometrics CH-250, Phoenix, AZ) as well as triple- and quadruple-color filtersets for multicolor FISH (Chroma Technology, Bellows Falls, VT).

Initially, we had experimented with a triple wavelength bandpass filter set to detect bound probes in red, green and blue. The blue fluorescent signals of probes detected with AMCA-conjugated avidin (Vector, Burlingame, CA), however, were consistently weak, prohibiting DAPI counterstaining of chromosomes. Our four-color filter set allowed specific excitation of DAPI (blue fluorescence) and additional fluorochromes in up to three different wavelength intervals. Bound probes were detected in individual wavelength intervals centered around 520 nm (FITC, green), 600 nm (rhodamine, red) and 690 nm (Cy5, infrared).
The quadruple-color filter set allowed simultaneous counterstaining of the DNA with DAPI, thereby greatly facilitating the task of finding metaphase cells. The image acquisition system used a monochrome CCD camera and a computer controlled filter wheel to select the wavelength of excitation light, and fluorescence images were acquired individually through a multi-bandpass filter set. For display on RGB monitors and in figures contained in this paper, we assigned the color purple to CY5 signals (red excitation) and show the blue fluorescence from DAPI counterstain (UV excitation) in form of a gray-scale image.
RESULTS
Chromosome 10 was chosen to develop the method of staining “chromosomal rainbows” for cytogenetic analysis. On a normal metaphase chromosome 10, our scheme began (from pter to qter) with a probe visualized in red close to the telomeric region of the short arm of chromosome 10, followed proximally by a probe detected in infrared, next a probe seen in green, and then one in yellow. The next probe, detected in red, started a new cycle of colors (Fig. 2A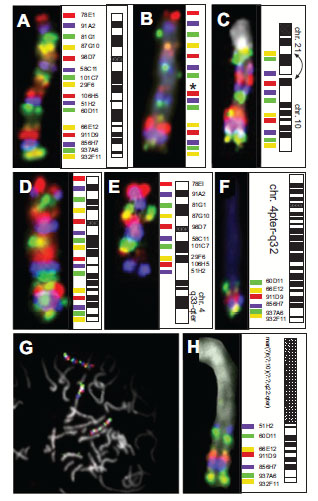 ), creating a pattern of a chromosome-specific 'rainbow'. Probes derived from YAC clones gave stronger signals than probes prepared from P1 clones (Table 1), which provided further clues for the identification of marker chromosomes (see below).
), creating a pattern of a chromosome-specific 'rainbow'. Probes derived from YAC clones gave stronger signals than probes prepared from P1 clones (Table 1), which provided further clues for the identification of marker chromosomes (see below).

Applying the technique to our model system of TPC-1 metaphase spreads, all previously described aberrations involving chromosome 10 [32, 36] could be detected. One homologue of chromosome 10 showed a deletion of locus D10S170 (probe 29F6) (Fig. 2B). The complex translocation involving chromosomes 1, 10 and 21 was detected in all metaphases analyzed (N=12). The breakpoints along chromosome 10, the paracentric inversion and the orientation of the rearranged parts of chromosome 10 could be described easily based on the sequence of colored hybridization signals. The entire p-arm, the centromere and band 10q11 with the breakpoint being distal to probe 58C11, was found reannealed to chromosome 1 fusing to the breakpoint on 1q with its p-terminus in accordance with a previous analysis using a different set of hybridization probes [32]. The remainder of chromosome 10, i.e., part of the long arm, showed a paracentric inversion which was detected by the inverted location of probe 58C11 (purple) and probe 29F6 (yellow) (Fig. 2C, arrows). Clone 58C11 contained the 3'-end of the ret gene (unpublished). One of the two breakpoints in the paracentric inv(10) (also called 'PTC1') fusing the 3’-end of the proto-oncogene RET to the expressed sequence D10S170 maps in intron 11 of the RET gene [4, 31], revealing that the entire signal from 58C11 (purple) was translocated.
One single experiment allowed us to map the breakpoint in the case of a reciprocal translocation t(4;10)(q33;q23.2) to the interval flanked by markers D10S507 and D10S541. The rainbow hybridization showed a normal chromosome 10 (Fig. 2D) in the presence a derivative chromosome 10 with chromosome 4-specific material fused to chromosome 10 distal of D10S607 (probe 51H2, purple) (Fig. 2E). These hybridizations used only 15 of the 16 probes, and did not include the probe for D10S170 (clone 29F6). The derivative chromosome 4 was found fused to material from the long arm of chromosome 10 distal of D10S541 (probe 60D11, green) (Fig. 2F). In this case, chromosomal rainbow hybridization indicated neither deletions nor chromosomal inversions.
Using the CR10 rainbow technique, a previously unknown marker chromosome could be identified and described in the follicular thyroid carcinoma cell line FTC-236 [44]. Metaphase spreads showed two apparently normal copies of chromosome 10 in the presence of a marker chromosome, which contained additional chromosome 10 material (Fig. 2G, arrow). The intensity and sequence of colored signals indicated that this marker carried part of the 10q arm (10q22.3-qter). The breakpoint maps between markers D10S537 (probe 106H5), which is absent from the marker chromosome, and D10S607 (probe 51H2, purple), which was found on this derivative chromosome (Fig. 2H).
For verification, overexpression of RET in TPC-1 cells could rapidly be demonstrated by hybridization of a biotinylated cDNA probe (Fig. 3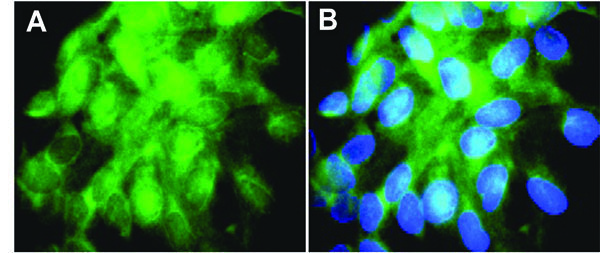 ). The RET specific transcripts (green) reported to be absent in normal thyroid specimens and adenomas [45] were detected in essentially every TPC-1 cell.
). The RET specific transcripts (green) reported to be absent in normal thyroid specimens and adenomas [45] were detected in essentially every TPC-1 cell.

DISCUSSION AND CONCLUSION
To facilitate screening of tumor specimens for aberrations involving specific chromosomes, we aimed to develop a technique which enabled us to detect inter- as well as intra-chromosomal rearrangements that were beyond the sensitivity of FISH with WCP probes. The availability of locus-specific probes and novel reporter molecules in combination with advances in optical filter design prompted the development of what we termed 'chromosomal rainbow' hybridization. Multicolor FISH with simultaneous hybridization of chromosome-specific probes had been proposed earlier [11, 20], but as we illustrate through several examples in this paper, a densely hybridizing panel of probes is required to achieve sufficient resolution and sensitivity to detect rearrangements.
In our CR10 probe panel, hybridization quality and efficiency of the probes was consistently high. We incorporated as many P1 clones as possible into the panel since these are generally easier to propagate and more stable than YAC clones [34]. With our spread of probes we could essentially detect any rearrangement involving more than 6-8 Mbp, but would probably miss some of the smaller rearrangements, when they occurred between two probes. When small interstitial deletions are suspected, we recommend that the analysis is performed on interphase nuclei, because the chromatin is less condensed so that closely-spaced probes show reduced overlap.
Chromosome 10 was chosen for the establishment of this technique because of its significance in thyroid cancer [5, 8, 23, 31, 46, 47]. Aberrations often involve intrachromosomal rearrangements, e.g., inversions, which cannot be detected by chromosome painting [3, 48]. The use of this chromosome 10-specific probe set, however, will not be limited to thyroid tumors, because rearrangements of chromosome 10 have also been observed frequently in other tumors, such as cancers of the prostate and brain. An increasing number of physically mapped probe collections, such as BAC or YAC libraries [33, 49-51] will facilitate the construction of additional rainbow hybridization panels.
Additional types of DNA probes might be useful and should be considered in such rainbow sets. Deng, et al., for example, described the development of chromosome band-specific probes prepared by microdissection, PCR and cloning [52]. This technique was further developed by Guan, et al. to the point, where essential coverage of chromosome 6 was achieved by hybridization of 14 subregion-specific probe libraries [53]. While utility of these probes is limited to metaphase chromosome analysis and interphase signals cannot be evaluated, they appear less useful for generating a rainbow hybridization panel due to low resolution. Recently, Liehr, et al. demonstrated that a combination of YAC/BAC’s and band-specific painting probe prepared by chromosome microdissection can overcome gaps in the BAC/YAC coverage and increase the banding effect [21]. Even more remarkable, Iourov and colleagues extended the use of ‘multicolor banding’ to the analysis of interphase cell nuclei in cases of mosaic neural aneuploidy [54, 55]. However, concerns relate to probe propagation, distribution and quality control: single clones have no issues with complexity and are easier to maintain, propagate and quality control than high complexity chromosome painting libraries. Furthermore, locus-specific probes can be tailored such that several probes hybridize within the same chromosome band.
The present study focuses solely on rearrangements involving chromosome 10. However, additional susceptibility loci for Familial Non-Medullary Thyroid Cancer (FNMTC) have been recently identified in classic isolated cases of FNMTC (1q21, 6q22, 8p23.1-p22, and 8q24) [47, 56]. Extrapolating our probe spacing to chromosome 6, we would apply 25 probes to cover the entire chromosome 6, whereas Guan, et al. reported 14 useful probe libraries derived from the dissection of individual chromosome bands [53]. Using BAC clones, probe coverage can easily be increased to achieve a sub-Mbp resolution, while probe pooling strategies minimize the steps to prepare the FISH probes without sacrificing sensitivity [22, 51, 57, 58].
In its present state of development, rainbow hybridizations allow staining of only one single chromosome for unambiguous mapping of breakpoints and marker chromosome identification. This limitation might be overcome by using additional reporter molecules and/or observation in multiple fluorescence wavelength intervals. Also, more sophisticated equipment such as the spectrofluorometer approach utilized in 'Spectral Karyotyping' or ‘Spectral Imaging’ [59, 60] might extend the useful range of this promising technique.
Nevertheless, results presented here underline the immediate utility of the ‘chromosomal rainbow’ technique. The observation of a breakpoint at 10q22-23 in FTC-236, for example, may document a non-random aberration in follicular thyroid carcinomas, similar to studies implicating this region in thyroid adenomas [61]. The breakpoint in our t(4;10) carrier, on the other hand, mapped close to the marker D10S541 which appears to fall in a rather unstable genomic region reported to be rearranged in numerous human tumors such as gliomas, breast and prostate cancer [62, 63]. While the carrier of the balanced translocation showed no disease symptoms other than reproductive problems, it is likely that allelic loss leading to inactivation of tumor suppressor genes in thyroid adenomas [61] may also be found in FTC-236 and related cell lines. Further studies of loss of heterozygosity (LOH) or FISH as says using gene- and locus-specific probes in the interval D10S541-D10S607 (i.e., PTEN or MMCA1 and D10S579, D10S1735, D10S1739, respectively) will help to evaluate possible alterations of these loci in thyroid tumors. We prefer FISH studies with probes densely distributed in the region of interest, since, unlike LOH probes, all FISH probes are informative and allelic loss will become evident immediately by counting the number of hybridization signals for any locus being studied.
In sporadic as well as radiation-induced PTC, ret rearrangements are the most commonly observed abnormality [5, 46], often termed ‘RET/PTC rearrangement’. It is interesting to note that all known translocation partners of the ret locus are genes which are constitutively expressed in thyroid follicular cells. In addition to driving the expression of the chimeric RET/PTC oncogene, these partners also provide a dimerization domain essential for ligand-independent activation of the ret tyrosine kinase [46]. As Tong, et al. could demonstrate, the leucine zipper-mediated dimerization is essential for the PTC1 oncogenic activity and tumor formation in mice [7, 64].
Unlike breakpoint-spanning probe contigs described earlier, which are testing only the integrity of the ret locus [32, 36], the novel rainbow hybridization scheme allows simultaneous delineation of intra-chromosomal rearrangements, thus expediting the process of identifying additional, novel RET/PTC translocation partners and with it, potential new targets for therapeutic intervention.
Although our examples illustrate the utility of the method to delineate breakpoints on the long arm of chromosome 10, the same set of probes and hybridization protocol can find immediate application in screening for translocations involving the short arm, which harbors a number of tumor-related genes such as the Kruppel-like transcription factor tumor suppressor gene KLF6 [65] or the AF10 gene involved in acute T-cell lymphoblastic leukemia [66].
In conclusion, we present here a novel high-resolution FISH based technique that is immediately applicable, as demonstrated by our successful validation experiments, but that will also form the basis for a variety of future developments, variations and refinements in rapid and specific scanning for inter- and intra-chromosomal rearrangements.
CONFLICT OF INTEREST
The authors declare that they do not have a conflict of interest.
DISCLAIMER
This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor The Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or The Regents of the University of California. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof, or The Regents of the University of California.
ACKNOWLEDGEMENTS
We thank Drs. T. Tanaka, S. Jhiang and P. Goretzky for supplying cells and cell lines. This work was supported in parts by a grant from the Director, Office of Energy Research, Office of Health and Environmental Research, U.S. Department of Energy, under contract DE-AC02-05CH11231, the Leonard Rosenman Fund, the Medical Research Service of the Veterans Affairs Medical Center, San Francisco, and NIH grants CA80792, HD45736, CA123370 and CA136685 and part of the project 7 of the Research Consortium Tumor Stem Cells funded by the Deutsche Krebshilfe (German Cancer Aid). Ideograms were kindly provided by D. Adler, Ph.D., Dept. of Pathology, Univ. Washington. We acknowledge the support from staff at Reprogenetics providing metaphase spreads and mapping data. Figures 2A-C are reproduced from Zitzelsberger, O’Brien and Weier: Multi-color FISH-based techniques for the detection of inter- and intrachromosomal rearrangements. Springer Laboratory Manual – FISH Technology, Edited by Rautenstrauss and Liehr. Springer Verlag, Heidelberg, 2002.